Linfoma cutáneo de celular T, micosis fungoides, reporte de caso y revisión del tema
Responde:
Dr. Cesar Daniel Alonso Bello*, Dra. Mayra Matías Carmona*, Dra. Paola Yolotzin Valenzuela Torres** Dra. Verónica Mena Zepeda, ****Dra. Adriana Machado Chavelas, *****Dr. José Manuel Conde Mercado****
*Médicos residentes en curso de posgrado en Medicina Interna.
*Médico residente en curso de posgrado en Anatomía Patológica
***Médico especialista en hematología, adscrita al servicio de hematología
****Médico especialista en Dermatología, adscrita al servicio de dermatología
*****Profesor titular del curso de posgrado de Medicina Interna
Introducción:
El linfoma de células T es el término actual que se utiliza para enfermedades caracterizadas por acumulación primaria de células T malignas en la piel. Los pacientes con las dos variantes predominantes: Micosis Fungoides (MF) y Síndrome de Sézary, característicamente desarrollan inmunodeficiencia severa durante la progresión de la enfermedad y por esto pacientes con enfermedad avanzada , mueren por infecciones y no por la actividad de la enfermedad. El linfoma cutáneo de células T (LCCT) representa un complejo de patologías con variadas manifestaciones, cursos clínicos y consideraciones terapéuticas. La MF es la forma más frecuente de LCCT, hay mucho sin conocer de la inmunología básica y biología molecular que de otras variantes.
Reporte de caso
En 2013 se presenta masculino de 60 años de edad, originario y residente del Distrito Federal,, escolaridad primaria incompleta, comerciante informal de artículos usados, casado, católico, peso 68 kilogramos y talla 1.65 metros.
Antecedentes personales no patológicos : Habita en casa de lámina, cuenta con agua, luz, drenaje, cohabita con 11 personas más. Tabaquismo desde los 15 años a razón de 12 cigarrillos diarios, índice tabáquico de 9. Etilismo desde los 15 años, 1 vez a la semana, no especifica tipo de bebida y cantidad. Niega toxicomanías, tatuajes, perforaciones, exposición a benceno, solventes, hidrocarburos, radón o radiación.
Antecedentes personales patológicos: Neurocisticercosis a los 20 años de edad, presentando como secuela convulsiones tónico – clónicas generalizadas, en tratamiento con carbamazepina 200 mg cada 8 horas, con adecuado apego al mismo.
Padecimiento actual: Inicia su padecimiento hace 11 años al presentar dermatosis diseminada a cabeza, cuello con tendencia la generalización, respetando palmas y plantas. De aspecto monomorfo polilesional constituido por 4 neoformaciones exofíticas de diversos tamaños, consistencia firme, ulceradas con costra en la superficie. Placas infiltradas de diferentes tamaños y formas, con escama en la superficie, borde infiltrado, algunas hipopigmentadas. Evolución crónica, pruriginosa y dolorosa. Acude a consulta a centro dermatológico donde se diagnostica MF, se inicia tratamiento con fototerapia en 51 sesiones, dexametasona intralesional en dos ocasiones y metotrexate 7.5 miligramos 2 veces por semana por 6 semanas.
Es referido de otra institución hospitalaria con resultados de biopsia de piel. Descripción: Los cortes muestran una epidermis con escasa capa cornea y focos de queratosis acantosis regular a moderada a expensas de los procesos interpapilares los cuales se anastomosan en algunas zonas atrapando papilas en dermis superficial se observan moderados infiltrados linfocitarios perivasculares, algunos linfocitos presentan núcleos hipercromáticos , invaden la epidermis y tienden a formar microabscesos de Pautrier . El resto del corte sin alteraciones, diagnóstico histológico: sugiere MF. Se indica tratamiento con crema emoliente, analgésico de tipo opiáceo, se solicita realización de Tomografía Axial Computarizada de cabeza, cuello y tórax y frotis de sangre periférica: Se observa engrosamiento, aumento de la densidad y presencia de lesiones nodulares, en los tejidos blandos, en la región temporo-occipital y mandibular izquierdas. Estas áreas presentan densidades, de hasta 50 unidades H, sin cambios con la administración de medios de contraste.
El parénquima cerebral, tiene leve aumento de la profundidad de los surcos, y la amplitud del espacio subaracnoideo, con adecuada diferenciación entre la sustancia gris y blanca, sin evidencia de lesiones ocupativas. Ambas parótidas presentan crecimientos ganglionares. Con crecimientos de tipo ganglionar, en los niveles 1 2 y 3 izquierdo, el de mayor tamaño en este último, con diámetro de 15.7 x10mm. Se observan crecimientos ganglionares, supra e infraclaviculares, de hasta 19.7 X 15 mm para el mayor del lado izquierdo, en las regiones axilares se observan ganglios que conservan su hilio graso, de hasta 11 mm. Diagnóstico de linfoma cutáneo en estadio tumoral. Frotis de sangre periférica linfocitos 25 000 células, monocitos 3000, eosinofilos 1, neutrófilo 71. Continua tratamiento con crema emoliente, hidroxicina 10 miligramos cada 24 horas y analgésico de tipo opioide.
Es ingresado al servicio de hematología, para realización de aspirado de médula ósea.
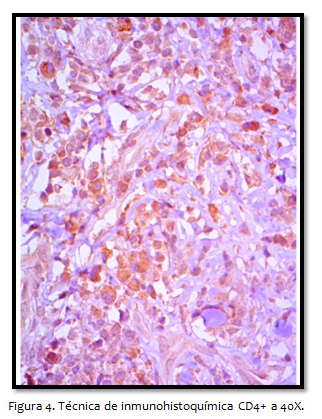
Descripción: médula ósea normocelular, con depósito de hemosiderina, negativo a malignidad. Por lo cual se inicia quimioterapia, 6 ciclos con ciclofosfamida 1275 mg, doxorrubicina 76 mg, vincristina 2 mg, prednisona 100mg sin alcanzar respuesta parcial. En cuatro meses se programa nueva biopsia de piel cabelluda, reportando LCCT compatible con MF componente foliculotrópico acentuado. En corte teñido con hematoxilina y eosina se observa infiltrado linfoide con atipia acentuada, y epidermotropismo difuso, prominente en segmento s foliculares profundos. El inmunofenotipo avala MF, por el marcaje predominante para linfocitos T (CD3 y CD4 positivos) con depleción de C5 C7 Y CD8. Ki67 40%. ELISA para virus VIH 1 y 2 y hepatitis C sin reactividad.
Se decide inicio de esquema de clonixinato de lisina + valproato de magnesio sin respuesta. Tres meses más tarde presenta como complicación infección de tejidos blandos de piel cabelluda, tratado de forma intrahospitalaria Se decide inicio de quimioterapia DHAP, el paciente no acepta. Iniciode metotrexate semanal 30mg. prednisona 25 mg y ácido fólico. Amitriptilina
12.5 mg/día, tramadol 50 mg/día, gabapentina 600 mg/día para manejo analgésico.
Epidemiología
La MF es un tipo de linfoma no Hodgkin extranodal relativamente raro con una incidencia de aproximadamente 0.36 por cada 100,000 individuos al año. En Estados Unidos de América se diagnostican 2000 nuevos casos anuales, tiene una mediana de edad de 57 años, es más frecuente en hombres que en mujeres en relación 2:1. Agentes infecciosos, exposición ocupacional y mutaciones genéticas han sido evaluados como factores etiológicos sin embargo en ninguno se ha comprobado una clara asociación con la enfermedad. El 97% de pacientes con estados tardíos de MF o Síndrome de Sézary son seropositivos para citomegalovirus en contraparte con los donadores de médula ósea que son seropositivos en 57%. Los estadios tempranos de la enfermedad tienen una media de sobrevida de 20 años y estadios avanzados de 5 años.
Inmunopatogénesis
En muchas leucemias y linfomas de células T no cutáneos existen asociaciones claras con mutaciones y anormalidades cromosómicas específicas en los cuales la expresión de oncogenes o inactivación de genes supresores de tumor resultan en expansión clonal, sin embargo no existe una asociación directa con los LCCT. Se ha observado que mutaciones en p53, gen regulador del ciclo celular están asociadas con progresión de la enfermedad, de igual manera las deleciones en 1p, 17p, 10q, 19 y adición en 4q, 18 y 17q. Después de la estimulación mediada por antígeno bajo condiciones fisiológicas, la eliminación de la mayoría de células T activadas es esencial para prevenir proliferación celular excesiva (como en leucemia y linfoma) y en la sobreexpresión de células T mediadas por daño tisular (como en estados autoinmunes e inflamatorios). La activación de apoptosis por CD 95 y Fas ligando provee una vía alterna para la eliminación de estas células, existen otras vías de liberación de moléculas de daño celular como perforina, granzima y linfocitos T citotóxicos, por lo tanto no es de sorprender que en la MF se muestre una disminución en la expresión de Fas con el avance de la enfermedad. Se han identificado mutaciones en Fas en esta enfermedad, la mayoría dando como resultado un receptor no funcionante. Paradójicamente las células T malignas pueden expresar Fas ligando para eliminar células T CD8 antitumorales.
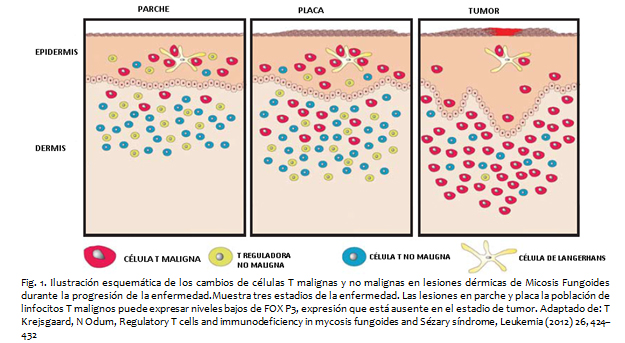
La idea de un subgrupo de células T inmunosupresoras ha sido de gran interés científico desde 1970. Estudios han comprobado que células T CD4+CD25+ previenen autoinmunidad en estudios con ratones y humanos. Las células T reguladoras se caracterizan por alta expresión de CD25+ y el factor de transcripción Forkhead Box P3 (FOXP3), cuya falta de expresión es causa de autoinmunidad en humanos por la falta de generación de linfocitos T reguladores funcionales por tal razón ha sido llamado el “regulador maestro” de estos linfocitos.
Aunque FOXP3 define la expresión de CD4+CD25+, la sola expresión de este factor no es determinante para conferir la función a los linfocitos T. La evidencia actual sugiere que los linfocitos T reguladores pueden dividirse en dos grupos, linfocitos T reguladores que se desarrollan en el timo y linfocitos T reguladores que se desarrollan en células tronco CD4+CD25- de activación periférica con presencia de factor de crecimiento tumoral beta (TGF-ß) e interleucina 2. A pesar de su origen diferente sus funciones periféricas parecen ser las mismas. La identificación de linfocitos T reguladores CD4+CD25+FOXP3+ revive la teoría de que los LCCT son derivados de un subgrupo de células T inmunosupresoras y esto explicaría la naturaleza inmunosupresora de la enfermedad.
Clasificación
A diferencia de otras patologías linfoproliferativas en que los hallazgos citogenéticos y de laboratorio juegan un rol principal en la estratificación de riesgo la estadificación TNMB (tumor, nódulo, metástasis, sangre) otorga un importante factor pronóstico en la MF y es la base para el tratamiento.
La estadificación actual y recomendación diagnóstica no requiere biopsia de nodos linfáticos clínicamente normales, sin embargo se recomienda una biopsia escisional o algún nodo clínicamente anormal (mayor o igual a 1.5 cm de diámetro) que preferentemente no tenga drenaje a una porción de piel involucrada
Clasificación TNMB de la MF
T: Piel
T0: Lesión clínica o histopatológicamente sospechosa
T1: Lesiones diagnósticas que afectan <10% de la superficie corporal
T2: Lesiones diagnósticas que afectan >10% de la superficie corporal
T3: Lesiones tuberosas o tumorales
T4: Eritrodermia generalizada
N: Ganglios linfáticos
N0: Sin ganglios linfáticos palpables o histopatológicamente no afectados
N1: Ganglios linfáticos palpables pero histopatológicamente negativos
N2: Sin ganglios linfáticos palpables pero histopatológicamente afectados
N3: Ganglios linfáticos palpables e histopatológicamente afectados
B: Sangre periférica
B0: sin células de MF en sangre periférica
B1: Más del 20% de células de MF en sangre periférica o más del 5% + clon T circulante
B2: más de 1000 células de MF/ mm 3 ó CD4/CD8 > 10 clon de linfocitos T circulantes demostrado por PCR o Southerblot
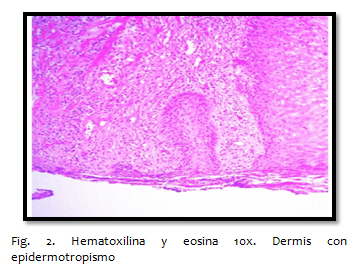
Los hallazgos histopatológicos varían de acuerdo a la etapa evolutiva de la enfermedad, en fases iniciales se caracterizan por placas con infiltrado linfocitario parcheado en la dermis superficial, con distribución perivascular, sin atipia nuclear, con tendencia de migración linfocítica hacia la epidermis por lo cual aparecen linfocitos salpicados entre los queratinocitos basales con alineación a lo largo de la unión dermo-epidérmica con cambios muy sutiles, lo que hace difícil el diagnóstico certero, por lo cual ante la sospecha se requiere un seguimiento con toma de biopsias. En la fase de placa , se observa un infiltrado linfocitario denso, dispuesto en banda a lo largo de la dermis papilar, con tendencia a agruparse constituyendo lo que se conoce como microabscesos de Pautrier, mismos que se caracterizan por linfocitos T CD4+ alrededor de una célula de Langerhans. Estos linfocitos intradérmicos se caracterizan por tener un núcleo grande, hipercromático, se pueden observar núcleos atípicos convolucionados y cerebriformes. En la fase tumoral el infiltrado es más denso, siendo nodular o difuso, afecta todo el espesor de la dermis con extensión frecuente hacia la hipodermis, las células se caracterizan por tener un núcleo muy grande, anaplásico y atípico, lo que implica mal pronóstico.
Respecto a la inmunohistoquímica, los linfocitos neoplásicos de la MF son T cooperadores, expresan los siguientes marcadores: pan-T, CD2, CD3, CD5, CD4, CD45RO, raramente expresan CD8 correspondiente a citotóxicos, además expresan proteínas citotóxicas como TIA-1 y granzima B. La fase tumoral se caracteriza por el cambio morfológico hacia células anaplásicas y grandes, que suele ir acompañada de perdida de marcadores de linfocitos T y la aparición de marcadores aberrantes. En resumen la micosis fungoide se caracteriza por el tropismo por los folículos y las glándulas sudoríparas, infiltración de linfocitos T en dermis y epidermis, linfocitos alineados en la membrana basal y microabscesos de Pautrier.
Diagnóstico
La valoración de este tipo de pacientes requiere una historia clínica y exploración física minuciosas, con especial atención a la presencia de adenopatías o de hepato-esplenomegali A como de exploraciones complementarias como biometría hemática, química sanguínea, pruebas de función hepática, frotis de sangre periférica, estudio histopatológico que incluya inmunohistoquímica, así como estudios de imagen como radiografía de tórax y TC de tórax, abdomen y pelvis. No se recomienda de manera rutinaria la biopsia de médula ósea, a excepción de pacientes que se encuentren en estadios tumorales, en casos de sospecha de enfermedad visceral o con fenotipo citotóxico (CD8+, granzima, TIA-1). La MF se considera un linfoma indolente originado en los linfocitos CD4+ recirculantes que presentan una gran afinidad por la piel, especialmente la epidermis (epidermotropismo), sin embargo se conoce como la gran simuladora debido a que las lesiones iniciales pueden presentarse eccematosas, liquenoides, psoriasiformes, etcétera; lo que obliga a descartar dermatosis inflamatorias, sumado a esto los hallazgos histopatológicos poco característicos hacen de difícil diagnóstico dicha enfermedad en fases iniciales, por lo cual resulta fundamental establecer una correlación entre la clínica e histopatología. Clínicamente de manera inicial se presentan máculas eritematosas persistentes y descamativas, en la fase de placa las lesiones se hacen gradualmente más elevadas e infiltradas al tacto, se pueden desarrollar a partir de lesiones maculares o aparecer “de novo” sobre piel previamente sana. Las placas son morfológicamente diferente de acuerdo al tamaño y forma, puede presentar regresión espontánea, permanecer estables o evolucionar a nódulos o tumores. Los tumores pueden desarrollarse a partir de placas o de piel normal.
Frecuentemente tanto las placas como los tumores pueden presentar necrosis y ulceración, otra manifestación común es la eritrodermia exfoliativa.
En la fase de placas se puede observar la presencia de linfadenoptías reactivas superficiales, mientras que en la fase tumoral se palpan linfadenopatías profundas específicas, a veces asociadas a lesiones viscerales.
La obtención de una biopsia representativa es esencial para valorar las características estructurales, se recomiendan biopsias amplias (en huso). Inicialmente se pueden requerir numerosas biopsias debido a los cambios poco específicos, hasta el característico infiltrado en banda formado por células mononucleares de tamaño pequeño o intermedio con núcleo cerebriforme e hipercromático, así como abscesos de Pautrier. En las fases tardías no suele observarse epidermotropismo. El estudio inmunofenotípico de biopsias en fresco o incluidas en parafina con anticuerpos monoclonales dirigidos contra antígenos de superficie celular o citoplasmáticos permite caracterizar poblaciones celulares linfoides clonales.
La demostración de una proliferación linfoide monoclonal en casos en los que se sospecha el diagnóstico de un linfoma se realiza a través del reordenamiento de los genes del receptor de la célula T, mediante la técnica de reacción en cadena de polimerasa (PCR) con la correlación clinicopatológica. Sin embargo, el fenotipo predominante es CD3/CD4+, algunos expresan CD3/CD8+ presentando estos últimos un curso más agresivo. La mayor parte de los pacientes expresan antígenos pan-T como CD3 y CD5 así como CD2, es frecuentes la pérdida de alguno de ellos hasta en el 50-70%, sobre todo en las formas tumorales.
Tratamiento
El tratamiento depende de la fase evolutiva en la cual se encuentra, las características clínicas y de la estadificación; en fases iniciales el tratamiento de elección son los corticoides, con adecuada regresión de la lesiones, mismas que recidivan al suspenderlos.
Los distintos abordajes terapéuticos consiguen únicamente mejoría sintomática y clínica (remisión parcial o completa), actualmente no existe un tratamiento curativo. Sin embargo las remisiones completas son frecuentes, especialmente en estadios precoces.
TABLA 1. ALGORITMO DIAGNÓSTICO DE LINFOMA CUTÁNEO DE CÉLULAS T
Examen físico completo
-Exploración de piel
-Exploración de adenopatías palpables y hepato-esplenomegalia
-Clasificación TNM
Estudios bioquímicos
-Biometría hemática y frotis de sangre periférica (células de Sézary), química sanguínea y pruebas de función hepática, lactato deshidrogenasa, ß2 microglobulina, serología para citomegalovirus, virus de Epstein-Barr, virus linfotrófico humano tipo I y II, virus de inmunodeficiencia humana, virus de hepatitis C, virus herpes tipo 6
Estudio de inmunofenotipo en sangre periférica
Radiografía de tórax
Ultrasonografía abdominal
Tomografía computada de tórax, abdomen y pelvis.
Biopsia de adenopatías si están presentes
Biopsia de médula ósea
El tratamiento agresivo en fases precoces no ha demostrado mayor tiempo libre de la enfermedad, ni aumento en la sobrevida global. Actualmente se inicia de manera gradual con tratamientos conservadores; las estrategias terapéuticas se dividen en dos categorías: tratamiento con efecto cutáneo directo y tratamientos sistémicos.
Los tratamientos con efecto cutáneo directo son de primera elección en lesiones nodulares aisladas y placas, su eficacia disminuye en formas extensas (T2), con porcentajes de remisión completa (RC) menores al 50%. En estadios avanzados se requieren tratamientos sistémicos.
El primer grupo incluye fototerapia con PUVA (psoralenos conjuntamente con luz ultravioleta A), fototerapia con UVB (luz ultravioleta B), quimioterapia tópica con mecloretamina (mostaza nitrogenada) o carmustina (BCNU), gel de bexaroteno al 1% (retinoide), radiación cutánea total (baño de electrones) y radioterapia convencional para lesiones sintomáticas. Ningún estudio ha demostrado la superioridad de un tratamiento en concreto, la elección depende de la experiencia del médico.
TABLA 2. ESQUEMA TERAPEUTICO DE LOS LINFOMAS CUTÁNEOS DE CÉLULAS T
Denileukin-diftitox, bexaroteno, IL, monoclonales, TMO
ICE: irradiación corporal con electrones; IFN-a: interferón alfa; TMO: trasplante de médula ósea
Tratamientos con efecto cutáneo directo
Fototerapia. Eficaz para los estadios T1/T2, puede conseguir una RC del 65-95%, respuestas parciales (RP) en T3/T4. La fototerapia con UVB alcanza remisión completa en más del 80% de los casos en T1, respuesta limitada a partir de T2, una limitación habitual son las áreas de flexión, ya que reciben una menor irradiación.
Quimioterapia tópica . Opción en estadios precoces o como tratamiento adyuvante tras conseguir remisión con fototerapia. La aplicación de mecloretamina 0.01-0.02% o BCNU consigue resultados similares a la radiación UV. Se asocia a dermatitis por contacto y reacciones de hipersensibilidad, por lo cual se limita su aplicación, la aplicación continuada ocasiona leucopenia y atrofia cutánea con telangiectasias. El periodo de aplicaciones varía entre 2 y 12 meses.
Corticoides tópicos. En estadio T1 se ha observado RC 60% y RP 90%, eficacia limitada en estadios más avanzados.
Retinoides tópicos. El bexaroteno al 1% tiene eficacia limitada en T1-T2, su uso se ha extendido a lesiones en áreas de flexión, su aplicación 2 veces al día consigue una respuesta del 60%, está contraindicado en el embarazo.
Radioterapia. Los linfomas suelen ser radiosensibles; la radioterapia con electrones aplicada total (baño de electrones) es muy eficaz en estadios iniciales (RC >90% en estadios I y IIA), su radiación solo alcanza la dermis papilar, no se ha demostrado superioridad respecto a los otros tratamientos ya mencionados. Se utiliza en pacientes con afectación cutánea difusa o extensa por placas gruesas e infiltradas (estadio IB) o con múltiples tumores (IIB) y en lesiones refractarias. La pauta consiste en alcanzar una exposición acumulada de 36 Gy en 10 semanas. Una vez alcanzada la remisión, suele requerirse la prescripción de un tratamiento adyuvante, puesto que la recaída es factible.
Tratamiento sistémico
Incluyen diversas pautas de poliquimioterapia, fármacos citotóxicos y modificadores de la respuesta inmune.
Pautas de mono o poliquimioterapia. Se usa como tratamiento paliativo en casos de recaída, refractariedad o etapas avanzadas. En general los linfomas T son quimiorresistentes, solo se consiguen respuestas parciales y de corta duración. No hay estudios que avalen un aumento en la supervivencia, se utilizan fármacos alquilantes como la mecloretamina, la ciclofosfamida, el clorambucilo o metotrexato.
Monoterapia. El metotrexato se usa en casos avanzados, con administración de dosis semanales intramusculares o subcutáneas de 50mg durante 2-4 meses.
Poliquimioterapia. Empleada en enfermedad avanzada, refractaria o con afectación sistémica, se han utilizado multiples pautas como: CHOP (ciclofosfamida, vincristina, adriamicina, prednisona), CAVE (ciclofosfamida, adriamicina, vincristina, etopósido) o EPOCH (etopósido, vincristina, adriamicina, ciclofosfamida, prednisona). Se ha observado recaÍda posterior a un par de meses, no se ha demostrado aumento en la supervivencia.
Fotoféresis. Indicado en pacientes con enfermedad en sangre periférica (Sézary), con respuestas superiores al 50% en casos refractarios a otros tratamientos, con mayor efectividad en pacientes sin antecedente de quimioterapia previa.
Tratamientos inmunooduladores. Interferones. El objetivo es restablecer una respuesta inmunitaria antitumoral, se ha observado respuesta global del 80% en estadio I y II, pero RC menores al 25%. Su administración puede ser vía subcutánea, intralesional o intramuscular, se inicia con dosis de 3-5 millones de unidades 3-4 veces por semana o diariamente durante 6-9 meses. Se ha visto que existe una relación dosis respuesta, por lo que puede aumentarse la dosis. En casos con elevado riesgo de recaída se puede utilizar dosis de mantenimiento por años, con el riesgo de desarrollar enfermedades autoinmunes.
Retinoides. El mecanismo de acción se basa en su acción antiproliferativa promoviendo la apoptosis, reduciendo la producción de citosinas y su actividad antiinflamatoria. El
bexaroteno se administra en dosis de 300 mg/m2 diariamente por 3-5 meses, eficaz en el 50% de los casos, se asocia a hipertrigliceridemia e hipotiroidismo como efectos adversos.
Trasplante de médula ósea. En casos de evolución agresiva o refractarios, consigue una remisión clínica mayor y más duradera, controla el desarrollo de nuevas lesiones cutáneas.
Discusión.
El linfoma de células T primario cutáneo de tipo MF, es el tipo más común de éste grupo aproximadamente en un 50% de todos los linfomas cutáneos primarios. Se caracteriza por infiltrados de tamaño pequeño a mediano de linfocitos T con núcleo cerebriforme limitado a piel. La diseminación extracutanea ocurre solo en casos avanzados principalmente a ganglios linfáticos, hígado, bazo, pulmones y médula ósea. Este término de MF sólo debe ser usado para los casos clásicos caracterizados por la evolución y combinación de parches, placas y tumores con una distribución amplia y curso de progresión lenta de años incluso décadas.
La histología de las lesiones varía según el estadio de la enfermedad, en la etapa de parches presenta infiltrados liquenoides constituidos por linfocitos e histiocitos y células atípicas con núcleo cerebriforme confinadas a epidermis. En las placas el epidermotrofismo es más pronunciado con agrupamientros intraepidermicos conocidos como microabcesos de Pautrier. En estado Tumoral hay pérdida del epidermotrofismo con infiltrados difusos y tamaños variables de las células cerebriformes. Típicamente el fenotipo celular se encuentra CD2+, CD3+, TCR B+, CD5+, CD4+,CD8-, en casos poco frecuente CD8+ y de presentación pediátrica principalmente.
El principal factor pronóstico es la extensión de la enfermedad cutánea y extracutánea que refleja el estadio clínico. La edad mayor de 60 años y elevación de la deshidrogenasa láctica se consideran factores pronósticos adversos así como la transformación histológica con incremento de células blásticas en más del 25%.
Las lesiones de nuestro paciente son lesiones clásicas de MF, con respuesta pobre a tratamiento tópico, la evolución de la enfermedad en el paciente fue hacia el deterioro no se administró quimioterapia por no consentirse por lo tanto no fue posible la evaluación de la respuesta a tratamiento, meses posteriores al rechazo de la quimioterapia el paciente es hospitalizado nuevamente por
presentar absceso en cuello y fallece por choque séptico.
Bibliografía
• T Krejsgaard, N Odum, Regulatory T cells and immunodeficiency in mycosis fungoides and Sézary síndrome, Leukemia (2012) 26 , 424–432
• Girardi M,. Heald Peter, The Pathogenesis of Mycosis Fungoides, N Engl J Med 2004;350:1978-88
• Weiyun Ai,, Keegan T. Outcomes After Diagnosis of Mycosis Fungoides and Sézary Syndrome Before 30 Years of Age, JAMA Dermatol . 2014;150(7):709-715
• Wilcox R, Cutaneous T-cell lymphoma: 2014 Update on diagnosis, risk-stratification, and management, American Journal of Hematology, August 2014 Vol. 89, No. 8
• Duque V, Velasquez M, Linfoma cutáneo de células T, inmunopatogénesis Universidad de Antoquia Colombia diciembre 2011, vol 24, 402-414
• Gallardo F, Pujol R, Diagnóstico y tratamiento de linfomas cutáneos T primarios, servicio de dermatología Barcelona España, actas demosifiliogr 2004;95
• Willemze R, Kerl H, Sterry W, Berti E, et al. EORTC Classification for Primary Cutaneous Lymphoma: a proposal from the cutaneous lymphoma study group of the European Organization for Research and Treatment of Cancer. Blood 1997;90:354-71
• Slater D. The new World Health Organizaton classification of haematopoietic and lymphoid tumours: a dermatopathological perspective. Br J Dermatol 2002;147:633-9
• Gómez S, Pérez N. Micosis fungoide y síndrome de Sézary. Actas Dermosifiliogr 2001;92:193-206
• Latkowski, J. Heald P.,Strategies for Treating Cutaneous T-Cell Lymphoma., J Clin Aesthetic Dermatol. 2009;2(6):22–27
Documento sin título
Próximo tema: A definir .
Se encuentra abierta la recepción de consultas para el próximo tema.
Las preguntas recibidas se seleccionarán por su frecuencia y calidad y conformarán parte del cuestionario a responder en la pxóxima entrega.