|
Discusión del
caso clínico
Dra. María Cecilia Demaría
Voy a discutir el caso de una paciente mujer de 30 años de edad hipotiroidea, monorrena, hipertensa, con probable eclampsia que presentó microangiopatía trombótica (MAT) en el post parto, por lo cual requirió internación en unidad de cuidados intensivos con requerimiento de ARM y de plasmaféresis así como de pulsos de corticoides. La paciente es dada de alta continuando en hemodiálisis por IRC secundaria a MAT. Reingresa por cuadro de disminución de agudeza visual bilateral y paresia braquial derecha, con imágenes no concordantes en tomografía computada (TC) que evidencia un área hipodensa en hemisferio cerebeloso izquierdo, ni en resonancia magnética nuclear (RMI) que evidencia imagen hiperintensa en la misma localización compatible con colección hemática subaguda. La paciente durante la internación agrega síndrome febril, por lo cual comienza tratamiento antibiótico cubriendo foco urinario e infección asociada a catéter de hemodiálisis. En laboratorio presenta LDH elevada. Evoluciona normotensa y afebril, con mejoría del foco neurológico a las 36 hs de internación.
Cómo interrogantes para abordar mi discusión planteo:
1- ¿Cuál es la causa de la microangiopatía trombótica?
2- ¿Estamos ante MAT como complicación del postparto o fue desencadenada
por el mismo?
3- ¿Cuál es la causa de las manifestaciones neurológicas?
4- ¿Estamos frente a una reactivación de su MAT?
5- ¿Cómo interpretar el síndrome febril en este contexto?
6- ¿Presenta algún síndrome que explique los antecedentes y cuadro actual?
7- Conclusión.
Las microangiopatías trombóticas (MAT) son desórdenes oclusivos microvasculares que se caracterizan por la agregación sistémica o intrarrenal de plaquetas, trombocitopenia y lesiones mecánicas de los eritrocitos. Cuando hablamos de MAT nos referimos a 3 entidades, púrpura trombótica trombocitopénica (PTT), síndrome urémico hemolítico (SHU) y asociada al embarazo. En la PTT la agregación sistémica provoca isquemia sobre todo en cerebro y en otros órganos; y en el SHU los trombos fibrinoplaquetarios ocluyen predominantemente la circulación renal. Por lo cual la PTT y el SHU son 2 síndromes agudos en los que las anormalidades neurológicas orientan a PTT clásica y la insuficiencia renal a SHU.
La MAT se define con péntada de:
- Anemia hemolítica microangiopática: es el sello diagnóstico. Se define como hemólisis no inmune con coombs negativo. Hay fragmentación de glóbulos rojos que se evidencian como esquistocitos en frotis (Fig 1). Como hallazgos de laboratorio encontramos bilirrubina indirecta elevada, disminución de haptoglobina y LDH elevada. La LDH elevada da cuenta de la hemólisis y del daño tisular por isquemia sistémica.
- Trombocitopenia: generalmente con menos de 20.000 plaquetas por mm3.
- Signo-sintomatología neurológica: lo más común es la cefalea y la confusión, las convulsiones son una manifestación relativamente frecuente pero puede presentarse con focalidad como por ejemplo afasia transitoria, AIT, ACV.
- Alteración de la función renal: por agregación a nivel de circulación renal.
- Fiebre: es poco común en series de casos. Estamos obligados a pensar en otras causas cuando ésta aparece.
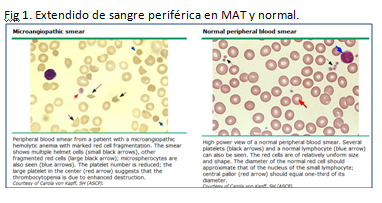
En la actualidad es suficiente la díada diagnóstica de anemia hemolítica microangiopática y trombocitopenia para comenzar tratamiento debido a la alta mortalidad que puede ser descendida desde 90 a 2% con tratamiento oportuno, recordemos que éste consta de plasmaféresis y corticoides sistémicos en casos de mala respuesta a la misma.
Como podemos ver, nuestra paciente presentó en el posparto todas las manifestaciones: anemia hemolítica con esquistocitos en frotis de sangre periférica, LDH elevada, coombs negativo, trombocitopenia, manifestaciones neurológicas e insuficiencia renal. Respecto de la diferencia entre ambos síndromes, en algunos pacientes, como la nuestra, se desarrollan alteraciones neurológicas graves e IRA, por lo cual son mejor descriptos con el término integral PTT- SHU. Sobe todo teniendo en cuenta que los cambios patológicos y el tratamiento no cambian sustancialmente.
En la bibliografía se describen indistintamente los diagnósticos diferenciales de esta entidad y las causas o condiciones predisponentes; y si bien la mayoría de los casos son idiopáticos, en muchos otros se pueden distinguir causas o estados asociados. Habiendo hecho una breve revisión del tema trataré de responder cuál es la causa en nuestra paciente (tabla 1).

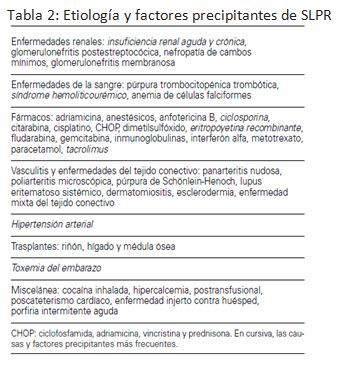
Cómo se puede apreciar en la tabla 1 hay causas que podemos descartar rápidamente. No presentó exposición a drogas tóxicas, ni en relación a quimioterapia por cáncer ni inmunosupresores. No presenta neoplasia diseminada sobre todo los carcinomas productores de mucina y no hubiese respondido a plasmaféresis. La quinina es un preparado de venta libre que es de uso común para tratamiento de calambres, puede desencadenar una MAT con un solo comprimido, incluso meses después del consumo; debe ser considerado debido a que produce MAT fundamentalmente con IRA y muchas veces su consumo pasa inadvertido debido a que hay bebidas de venta libre que la contienen, en nuestra paciente es una causa alejada ya que no sólo no contamos con el antecedente del consumo sino que tampoco se presenta con el cuadro compatible de dolor abdominal, nauseas, vómitos y diarrea. Otras causas pueden también ser descartadas: no consumió antiplaquetarios, tampoco se encontraba en tratamiento con ACO ni con valaciclovir, no es portadora de transplante halogénico ni infección por neumococo, aspergillus o CMV. No es portadora de HIV ya que contamos con serología negativa reciente. Tampoco presenta cuadro clínico ni fondo de ojo concordante con hipertensión maligna como causa desencadenante. Respecto de las enfermedades sistémicas autoinmunes el LES si bien puede cursar con alteraciones a nivel del SNC, no presenta clínica compatible, no reúne criterios y tenemos FAN y anti DNA negativo, es un diagnóstico alejado así como también la AR ya que no presentó artralgias ni clínica compatible y la afectación neurológica en general es a nivel del SNP. El SAF catastrófico puede cursar con MAT, pero nuestra paciente cuenta con anticuerpos anticardiolipinas negativos, no presenta antecedentes de trombosis arterial o venosa, ni de abortos y aunque considero que podrían repetirse los anticuerpos debido a que en cuadro agudo pueden resultar negativos, es un diagnóstico alejado. Otra de las causas frecuentes que debe ser tenida en cuenta por la insuficiencia renal aguda grave que llevó a la paciente a hemodiálisis es el cuadro asociado a diarrea disenteriforme causado por la toxina shiga producida por E Coli 0157 H7 que puede afectar a todas las edades, pero es más frecuente en niños, no presentó cuadro abdominal compatible ni diarrea, y la insuficiencia renal puede ser explicada por otras causas ; recordemos que hasta un 50% de los casos de PTT cursan con insuficiencia renal. Y antes de pasar a las causas más probables quiero mencionar a la CID, un cuadro que se produce por agregación de trombos de fibrina en lugar de plaquetas que en general se asocia a sepsis, preeclampsia y que cursa con trombocitopenia, disminución de fibrinógeno y de factor V y prolongación de tiempos de coagulación como presentó nuestra paciente. Creo que la paciente presentó un cuadro de CID como condición asociada a la MAT durante la internación previa.
La causa idiopática de la MAT es probable pero muchas veces ésta corresponde al déficit de ADAMTS 13. En condiciones fisiológicas el factor de von willebrand (FvW) es liberado por las células endoteliales en formas de muy alto peso molecular (ultragrandes), las cuales son escindidas por la proteasa ADAMTS 13 con el fin de limitar el tamaño y actividad. En la PTT la disminución cuantitativa o funcional de ADAMTS 13, resulta en un depósito y persistencia de éstas formas ultra grandes de FvW sobre las células endoteliales. Estas formas hiperactivas de FvW son capaces de reclutar plaquetas sobre la superficie celular. La unión de plaquetas al FvW determina una secuencia de eventos activación de agregación, formación de trombos endovenosos. El déficit de ADAMTS 13 puede ser congénito o adquirido. A favor de esta etiología, si bien muchas veces es por descarte, es la frecuencia respecto de las otras etiologías, la producción del cuadro de MAT y escasa mejoría luego de varios días posteriores del alumbramiento y la respuesta a la plasmaféresis. La plasmaféresis es efectiva en PTT y SHU, con ella se elimina del plasma del paciente el anticuerpo contra ADAMTS 13, por otra parte en el embarazo se produce un déficit fisiológico de ésta proteasa. Por lo tanto un cuadro de PTT pudo haber estado desencadenado por el embarazo o puerperio, situación que se observa en el 10 al 25 % de los casos. Es bien conocida la dificultad para realizar diagnóstico de PTT asociada a embarazo, sobre todo por el diagnóstico diferencial con eclampsia.
Entonces se abre el interrogante de si estamos frente a una MAT del embarazo. En el año 1954 Prichard describió cuadro de destrucción de eritrocitos, plaquetopenia y alteraciones hemostáticas en pacientes con eclampsia.
La preeclamsia-eclampsia es un síndrome que se caracteriza por hipertensión arterial (HTA) >140/90, proteinuria, luego de las 20 semanas de gestación y que se asocia a convulsiones en cuyo caso se denomina eclampsia. La base y sustento fisiopatogénico es la disfunción endotelial, y con ello mala perfusión sistémica y renal de manera temprana. Si no evoluciona de manera favorable se produce un estado trombogénico con depósito de fibrina en la microcirculación con destrucción eritrocitaria. Por lo cual estamos frente a anemia hemolítica y plaquetopenia constituyendo este cuadro la llamada MAT asociada al embarazo, cuyo tratamiento es el alumbramiento. Pero son bien conocidos los casos de preeclamsia-eclampia que se dan en el post parto y que como se describió pueden cursar como complicación con MAT. Se presume que un proceso evolutivo endotelial no desaparece luego del alumbramiento.
El interrogante de hasta cuanto tiempo luego del parto persiste la lesión endotelial, que no ha podido ser respondido aún en la actualidad. Ante esto planteo las siguientes posibilidades: en primer lugar, si bien no contamos con antecedente de proteinuria, si cuenta con las otras manifestaciones (HTA luego de las 20 semanas y convulsiones además de la evidencia de daño endotelial) de una eclampsia que se desarrolló post parto. Por lo cual creo que nuestra paciente pudo haber tenido un cuadro de eclampsia que complicó con MAT asociada, pero en contra de esto tenemos que se desarrolló y persistió varios días (más de los habituales que son 3) posteriores al alumbramiento, que respondió a plasmaféresis. O bien, en segundo lugar, pudo haber presentado un cuadro de eclampsia, y el postparto actúo como gatillo de una PTT como 2 entidades separadas, recordemos el déficit fisiológico de ADAMTS 13 durante el embarazo que sumado a las otras situaciones descriptas pueden justificarlo. O bien, y en tercer lugar pudo haberse tratado de una misma entidad, como podría ser la PTT por déficit de ADAMS 13 o idiopática y las convulsiones haber sido una manifestación neurológica de ésta sin haber presentado un cuadro de eclampsia, a favor de esto, así como de la situación anterior, la paciente respondió a plasmaféresis. También en este sentido y como ya mencioné el dosaje de ADAMTS 13 sería de utilidad para acercarnos al diagnóstico retrospectivo, ya que de esto dependería algunas conductas como por ejemplo desaconsejar nuevo embarazo.
Quisiera mencionar que la eclampsia es causa de muerte materna cuando se asocia a hemorragia cerebral y a MAT. Recordemos que la paciente en discusión presenta una RMI con imagen compatible con hemorragia subaguda. No se conoce con exactitud la fisiopatología de la hemorragia cerebral en la eclampsia pero las alteraciones hematológicas, la diátesis hemorrágica, el síndrome de fuga capilar y la falla orgánica múltiple en estados de MAT asociada podrían generar el terreno propicio para que ello se produzca. Si bien esta no es una asociación frecuente y cursa con una alta mortalidad, puede ser el caso de nuestra paciente.
Uno de los problemas principales de la paciente es la signosintomatología neurológica, que en primer lugar obliga a plantear el interrogante de si ambas manifestaciones son actuales y si existen causas que justifiquen ambas alteraciones o corresponden a diferentes causas. Y hay tres principales, de las cuales la neoplásica queda alejada en principio, debido a que no presenta síndrome de impregnación, cuadro clínico, laboratorio ni imágenes de SNC compatibles. La causa infecciosa si bien es un diagnóstico alejado lo retomará posteriormente en la discusión. Y dentro de la causa vascular, que es la más probable, voy a considerar 6 entidades:
- Accidente isquémico transitorio: inicialmente considerado debido a la ausencia de traducción estructural en métodos por imagen. Pero rápidamente queda descartado debido al tiempo de evolución de los síntomas que es más de 48 hs.
- Accidente cerebrovascular isquémico embólico: es atractiva la explicación de que las manifestaciones neurológicas no sean concordantes, debido a la alteración de más de un área cerebral, y las embolias múltiples la causa. Los datos que tenemos en contra son varios: no tenemos evidencia de cardiopatía embolígena, ya que contamos con ecocardiograma que no evidencia valvulopatía ni trombos intracavitarios con FSVI normal. No presenta antecedente, ni desarrollo actual de arritmias cardíacas. Por otro lado es extremadamente raro que los émbolos ocluyan de manera simultánea y bilateral la circulación retinal y la agudeza visual no se recupera rápidamente cuando es ésta la etiología. Por último los métodos por imágenes, sobre todo la RMI es altamente sensible y específica para isquemia aguda, y nuestra paciente no presenta evidencia de isquemia en la RMI. Por todo esto, lo considero un diagnóstico posible, ya que no lo puedo descartar.
- Embolias de colesterol: recordemos que esta entidad se describe de forma clásica como consecuencia de la emisión de cristales de colesterol procedentes de lesiones arterioescleróticas de distintos territorios orgánicos. En cuanto a la presentación clínica es variable, en función de los órganos afectados pudiendo observarse fiebre, mialgias, pancreatitis, claudicación, hemoptisis, insuficiencia renal, hemorragia digestiva, AIT, ACV, HTA, retinopatía con amaurosis fugáz. De ellos el riñón es el mejor marcador de afectación visceral. El diagnóstico se realiza con la biopsia del territorio afectado que en muchas ocasiones es la piel debido a la frecuente alteración de la misma y a nivel del fondo de ojo la observación de placas de hollensshort. Si bien nuestra paciente presentaba factores de riesgo como HTA, y dislipemia (en probable relación a mal control de hipotiroidismo), no tenemos evidencia de arterioesclerosis ni fue sometida a procedimientos endovasculares que precipiten el cuadro, al igual que en el ACV es extremadamente raro que se produzca en este contexto la oclusión simultánea de los pequeños vasos retinianos de forma bilateral. Por lo cual este diagnóstico lo considero alejado.
- Accidente cerebrovascular trombótico: considerando que la isquemia es causa del 85% de los ACV y que nuestra paciente presenta factores de riesgo tales como HTA, dislipemia, sedentarismo y un fondo de ojo que muestra máculas insatisfactorias, siendo este un reflejo de lo que ocurre en la circulación sistémica es un diagnóstico que estamos obligados a pensar. Ahora bien, todos los intentos de explicar un ACV trombótico como causa de las 2 manifestaciones clínicas neurológicas principales, se derrumban cuando repasamos el territorio arterial cerebral implicado. Y si considerásemos que sólo una de las manifestaciones es explicada por esta causa, es el síndrome retiniano que ocasiona neuritis óptica isquémica anterior, es irreversible y sólo en pocos casos es bilateral. El fondo de ojo siempre se encuentra alterado. Y el otro síndrome es el de la arteria cerebral media que puede presentarse como una monoparesia braquial pero que debería cursar con el resto de las manifestaciones a nivel sensitivo. A esto se suma que un método con alta sensibilidad y especificidad para detectar isquemia aguda como es la RMI, en nuestra paciente no muestra isquemia. Por lo cual lo considero un diagnóstico posible; y también considero posible que el foco motor sea manifestación de un foco neurológico previo que en este contexto de infección se haya puesto en evidencia.
- MAT reactivada: ya se argumentó previamente que la paciente no presenta reactivación, sobre todo teniendo en cuenta que no presenta plaquetopenia ni esquistocitos en frotis, que ya presenta un daño renal crónico por lo cual se encuentra en hemodiálisis siendo este un parámetro no tenido en cuanta para el diagnóstico. Con la salvedad de que en IRC la reactivación puede cursar con menor recuento de plaquetas y los esquistocitos pueden tardar en aparecer en el curso muy temprano de la enfermedad o muy cerca de un episodio previo de MATcomo en el caso de nuestra paciente. Recordemos que las manifestaciones neurológicas más frecuentes en MAT son cefalea y confusión pero que puede cursar con convulsiones, coma, ACV, AIT, y disminución de la agudeza visual. Lo llamativo de las alteraciones focales asociadas a MAT es cursan con estudios por imágenes normales o con imágenes compatibles con leucoencefalopatía posterior reversible.
Por lo tanto creo que es un cuadro ante el cual debemos estar alertas ya que se trata de manifestaciones neurológicas en paciente con antecedente de MAT, que presenta una LDH aumentada. Pese a este, lo considero un diagnóstico alejado.
- Síndrome de leucoencefalopatía posterior reversible (PRESS) previa: este cuadro fue descripto por primera vez en 1996 por Hinehez. Se trata de encefalopatía causada por afectación de la sustancia blanca cerebral, especialmente en áreas posteriores de los hemisferios cerebrales. De los 15 pacientes que se estudiaron inicialmente para describir el cuadro tres tenían eclampsia y todos HTA. En cuanto a la etiología son múltiples las causas, pero en coincidencia con nuestra paciente puede ser producida por PTT-SHU, eclampsia y transfusiones de sangre. Las dos teorías fisiopatológicas tratan de explicar que las lesiones cerebrales son secundarias a edema cerebral y éstas son: 1) Teoría de edema de origen vasogénico: sugiere que la elevación brusca de la presión arterial excede la capacidad de autorregulación cerebral, produce distensión y necrosis de arteriolas y capilares con el consiguiente edema vasogénico por trasudación capilar de líquido hacia el espacio intersticial. La rotura de la integridad de la barrera hematoencefálica a través de este mecanismo produce edema cerebral, hemorragias petequiales parenquimatosas y hasta infartos cerebrales. 2) Teoría del edema cerebral citotóxico: sugiere que el aumento brusco de la PA da lugar a un vasoespasmo y a fenómenos isquémicos con edema citotóxico y extracelular. Es posible que ambos tipos de edema contribuyan al cuadro. Estudios recientes demuestran que si las técnicas de difusión en diagnóstico por imágenes demuestran la progresión del edema vasogénico a citotóxico es indicativo de irreversibilidad e implica conversión a infarto e incluso hemorragia cerebral. En cuanto a las manifestaciones clínicas suele manifestarse como una encefalopatía de inicio agudo o subagudo; la sintomatología incluye cafalea, naúseas, vómitos y alteraciones de la visión (visión borrosa, hemianopsia y ceguera cortical) síntomas neurológicos focales y empeoramiento progresivo del nivel de conciencia. Las convulsiones pueden constituir el primer síntoma o aparecer tardíamente, en otros enfermos los primeros síntomas son somnoliencia, confusión, letargia, abulia, que alternan con episodios de excitación. El diagnóstico se realiza con la confirmación de las imágenes evidenciándose alteración de la señal en sustancia blanca, consecuencia del edema. La TC puede ser suficiente si muestra edema en forma de áreas de hipodensidad y en RMI lesiones iso o hipointensas en T1 e hiperintensa en T2. Las secuencias en FLAIR suprimen la señal en el área ventricular y el espacio subaracnoideo por lo que distinguen mejor las lesiones de la sustancia blanca y la corteza cerebral. Hay estudios que concluyen en que la hemorragia cerebral más PRESS está asociada a coagulopatía o diátesis hemorrágica. Aunque el PREES se asocia en general a un cuadro clínico benigno y la recuperación es lo esperado,el PRESS asociado a hemorragia tiene un curso clínico variable.
Es lógico pensar en predisposición a hemorragia en sitios de injuria aguda, sobre todo en pacientes con alteración de la hemostasia. Los infartos son más frecuentes en territorios limítrofes y el cerebelo es un sitio susceptible. Estas complicaciones dan focalidad neurológica transitoria o permanente (recordemos que la naturaleza fluctuante de la vasoconstricción determina que algunas lesiones no sean reversibles).
Habiendo hecho una descripción del cuadro, parece razonable pensar que las alteraciones neurológicas puedan corresponder a este cuadro. Si observamos el cuadro vemos que cuanta con muchas de las causas y factores precipitantes: IRA e IRC, PTT-SHU, HTA, toxemia del embarazo.
A favor de esta entidad tenemos evidencia en TAC previa de alteraciones en sustancia blanca compatibles con lesiones isquémicas múltiples, que podrían explicar a la la alteración parética como reactivación de lesión cerebral previa en este contexto de infección o bien se trata de un foco motor en regresión de aquel episodio. La lesión a nivel del centro semioval era de mayor tamaño que lo que se puede apreciar en la TC de esta internación. El control de la PA debió haber contribuido a la mejoría del cuadro.
Considero a este un diagnóstico muy probable en nuestra paciente.
Si la alteración motora corresponde a este cuadro, me pregunto ¿Hay más de una causa que explique éstas manifestaciones?
Me voy a referir a la disminución de la agudeza visual.
En primer lugar tratar de caracterizar la pérdida brusca de la visión ayuda a descartar de entrada varias etiologías: es binocular, dolorosa, con recuperación, con síntomas asociados de focalidad neurológica.
Dentro de las causas de pérdida brusca de la visión se encuentran las siguientes:

De la tabla descripta la mayoría de las causas quedan descartadas varias causas ya que la mayoría de éstas entidades cursa con alteraciones a nivel del fondo de ojo, otras presentan cuadro clínico incompatible, otras son unilaterales, otras son irreversibles, por lo cual sólo voy a considerar a la neuritis óptica. Y dentro de éstas a las neuritis ópticas retrobulbares, ya que en el caso de nuestra paciente cursó sin alteración del disco óptico.
En las neuritis retrobulbares (NORB) la lesión se sitúa por detrás del disco óptico. Cursan con disminución de agudeza visual, defecto pupilar aferente (DPA), pérdida del campo visual y fondo de ojo sin edema. Puede definirse como el tipo de neuritis en la que “el paciente no ve (disminución de agudeza visual) y el médico no ve (fondo de ojo normal)”. Los mecanismos por los cuales puede producirse la disminución de la agudeza visual en NORB son: desmielinización (alejado en este contexto), compresión (alejado ya que no presenta lesiones en SNC y la disminución sería insidiosa), infiltrativa (alejado la pérdida es progresiva), inflamatoria (no presenta evidencia de infección local ni de alteración inmunológica por clínica ni por laboratorio, por lo tanto este mecanismo también es alejado), traumática (descartado, no presenta antecedente de trauma) y el mecanismo más probable de producción es la isquemia. La Neuritis óptica isquémica posterior (PION) es una entidad que se caracteriza por afectación de las arterias ciliares posteriores cortas, dentro de las causas se encuentran las trombóticas y embólicas y por hipoperfusión. Dentro de los factores de riesgo se describen varios, que coinciden con los de nuestra paciente: HTA, dislipemia y el binomio anemia + IRC, entre otros. En cuanto a la clínica, tal y como se presentó en nuestra paciente cursa con pérdida de agudeza visual brusca, con posterior recuperación, con dolor, y puede ser bilateral, lo que podría explicar la ausencia del defecto pupilar aferente. Y también como en el caso de nuestra paciente con fondo de ojo normal. El diagnóstico de esta entidad es clínico. Por lo cual considero a este un diagnóstico muy probable.
No obstante esto, no quiero dejar de considerar a la neuritis óptica idiopática que es generalmente inflamatoria y afecta a adultos jóvenes, es de comienzo agudo, con dolor, puede ser bilateral , con fondo de ojo normal y un pronóstico excelente en el 95% de los casos, que es también un diagnóstico posible.
Ahora bien un capítulo aparte y no muy conocido lo constituye la disminución de la agudeza visual asociada a paciente en hemodiálisis.
La neuritis óptica asociada a IRC es una entidad respecto de la cual hay pocos trabajos publicados, y sólo 12 casos reportados. Se clasifica en tóxica urémica (alejado), por HTEC (descartado), infecciosa (muy alejado), isquémica (ya descripta previamente y muy probable) y por fármacos, que consideraré seguidamente. Por lo cual creo que es probable que en contexto de hipotensión intradiálisis se haya producido este cuadro.
No podemos dejar de considerar la medicación que venía realizando la paciente, y en particular voy a referirme al ácido tranexámico. Este es un potente inhibidor de la activación del plasminógeno, reduciendo la disolución de la fibrina hemostática por plasma, es decir es un agente inhibidor del plasminógeno. En presencia de ácido tranexámico los receptores de lisina de la plasmina para la fibrina están ocupados. Tiene indicaciones precisas como son prevención de hemorragia en hemofilia y en sangrados ginecológicos. La eliminación del fármaco es renal por lo cual requiere de ajuste de dosis y la administración en general no debe hacerse por más de 5 días. Está contraindicado en pacientes con defectos adquiridos de la visión, enfermedad tromboembólica venosa, antecedente de tromboembolismo en vena central de retina y en pacientes con riesgo de trombosis (hipercoagulabilidad). Las reacciones adversas son varias, pero quiero destacar las anormalidades visuales y alteraciones retinales, la trombosis y el tromboembolismo venoso y arterial así como obstrucción de la arteria y vena retinal. Teniendo en cuenta la farmacología tengo todo el derecho a pensar que las alteraciones visuales en nuestra paciente pueden estar en relación a este fármaco.
Encontré un solo reporte de caso de pérdida de agudeza visual en un hombre de 56 años que se encontraba en hemodiálisis que presentó disminución de la agudeza visual mientras se encontraba en tratamiento con ácido tranexámico por un cuadro de hemorragia digestiva; este paciente recupera la visión a las 48 hs, en asociación a la suspensión de dicho fármaco y presentó estudios normales (RMI, fondo de ojo). Son pocos los estudios sobre la discapacidad visual y ácido tranexámico, pero los que hay sugieren una fuerte asociación entre los pacientes en hemodiálisis y disminución de agudeza visual asociada al ácido tranexámico. Por todo esto, y considerando que la paciente recibió el fármaco por varios días más de lo que está indicado, que no se realizó ajuste de dosis según función renal, que no hay causas alternativas que expliquen la disminución de la agudeza visual, que presentaba contraindicaciones o precauciones para realizarlo, creo esta puede ser una causa de que justifique estos hallazgos.
¿Cómo interpretamos la fiebre en este contexto?
Como ya se mencionó la fiebre como signo de MAT es poco frecuente. En la mayoría de las series de casos no es un síntoma común de MAT y por el contrario, siempre que exista fiebre debe hacernos pensar en una causa infecciosa y descartarla.
En nuestra paciente los focos más probables son el urinario, ya que presentó un exámen de orina completa con leucocitos y piocitos, había estado sondada e institucionalizada previamente y por último para terminar de confirmar este diagnóstico se aisló en urocultivo bacilos Gram negativos.
Otro de los focos probables es la sepsis a catéter. Si bien la presentación no es de sepsis entendiéndola como SRIS asociado a infección documentada, queda demostrado que la infección asociada a catéter de hemodiálisis estuvo presente. Presento con hemocultivos y retrohemocultivos positivos como se mencionó en la presentación del caso, que no presentó registros febriles posteriores a la retirada del catéter. Y por último voy a considerar el cuadro de endocarditis infecciosa, debido a que nuestra paciente se presentó con fiebre asociada a un catéter venoso y manifestaciones neurológicas que pudieran ser explicadas por embolias sépticas. Las embolias en SNC deberían haberse traducido en imágenes tomográficas o de RMI. Claramente no se presentó con clínica compatible, ni signos de ICC, no reúne criterios de Duke ni de Duke modificados. Y por último el fondo de ojo no evidencia las manchas de roth (lesiones hemorrágicas exudativas y exudados en la retina), recordemos que le fondo de ojo de nuestra paciente a excepción de las alteraciones maculares es normal. Por lo cual creo que este diagnóstico es alejado.
Respecto de los síndromes que cursan con baja talla, hay mucho, pero el más compatible con los hallazgos de nuestra paciente es el cretinismo.
El hipotiroidismo puede ser congénito (cretinismo ) o adquirido (autoinmunización).
El cretinismo se produce por el fracaso del desarrollo embrionario de la glándula tiroides, es decir el 85% son esporádicos por disgenesia tiroidea y el 15% son hereditarias (causados por errores innatos de la síntesis de la hormona tiroidea o tiroxina). Cuando hablamos de disgenesia nos referimos a agenesia, hipoplasia o ectoplasia. Clínicamente se manifiesta en el nacimiento con ictericia, inactividad (sueño) anemia e hipotonía abdominal. En cuanto al desarrollo del SNC se manifiesta con retraso mental. Otras manifestaciones incluyen movimientos lentos y torpes, somnolencia, letargo. Baja talla y las características faciales: frente corta, ojos grandes, párpados arrugados, naríz corta y amplia, y macroglosia. Cabello seco, quebradizo, sin brillo, línea de implantación baja. Anemia con síndrome anémico y el déficit de vitamina A que pueden explicar la piel seca, gruesa y escamosa, como nuestra paciente. En cuanto a las determinaciones de laboratorio se presenta con TSH alta y T4 baja. Varias de las manifestaciones se corresponden con las características fenotípicas de la paciente. Hay muchas causas de baja talla y de éstas el diagnóstico más probables creo que es el de cretinismo. No encontré asociación de éste síndrome con las manifestaciones de microangiopatía trombótica.
Conclusión:
Creo que estamos ante una paciente mujer de 30 años, hipotiroidea, monorrena, IRC en hemodiálisis, HTA, dislipémica en probable relación a hipotoroidismo no controlado, que probablemente sea portadora de cretinismo. El cuadro clínico previo de MAT correspondió a microangiopatía asociada al embarazo y/o eclampsia, pero más probablemente corresponda a PTT desencadenada por el embarazo o post parto. La fiebre está perfectamente explicada por la infección urinaria y por la infección asociada a catéter de hemodiálisis. Y las manifestaciones neurológicas creo que están explicadas por causas diferentes, en cuanto al foco motor, probablemente corresponda a Síndrome de leucoencefalopatía posterior reversible previa (PRESS). Y en cuanto a la disminución de la agudeza visual lo más probable es que esté vinculada al uso de ácido tranexámico o bien corresponde a neuritis óptica isquémica posterior (PION). No presenta reactivación de la enfermedad de base (MAT) pero debemos estar alertas ya que la paciente presenta un fuerte antecedente reciente, manifestaciones neurológicas y LDH elevada.
El endotelio vascular, es la alteración que juega un rol protagónico en esta paciente.
Como conductas a seguir planteo :
- COMPLETAR TRATAMIENTO ANTIBIÓTICO AJUSTADO A SENSIBILIDAD.
- REGULACIÓN DE HORMONA TIROIDEA.
- PREVENCIÓN FACTORES DE RIESGO CARDIOVASCULAR.
- SEGUIMIENTO ESTRICTO Y DESACONSEJAR EMBARAZO.
- DOSAJE DE ADAMTS 13.
Bibliografía
- F. López-García a, F. Amorós-Martínez a, A.P. Sempereb. Síndrome de leucoencefalopatía posterior reversible. REV NEUROL 2004; 38 (3): 261-266.
- Rosalyn M. Aranas Æ ShyamPrabhakaranÆ Vivien H. Lee. Posterior Reversible Encephalopathy Syndrome Associated with Hemorrhage. Neurocrit Care (2009) 10:306–312.
- Fiona Costello RETROBULBAR OPTIC NEUROPATHIES. 2009, American Academy of Neurology. All rights reserved.
- Joel L. Moake, MD. MECANISMOS DE LA ENFERMEDAD. Microangiopatía trombótica. N Engl J Med 2002; 347:589-600.
- Omar Eguia-Ortega, Miguel A. Zambrano-Velarde, Leonardo Pazarin-Villaseñor Purpura TrombocitopénicaTrombótica Asociada a Embarazo: un Diagnostico Complejo. MedHosp 2013;1(2):24-26
- Enrique Gómez de la Fuente, Francisco Javier Vicente Martín, José Gregorio Álvarez Fernández. Embolismo por cristales de colesterol, con fracaso renal agudoo subagudo, diagnosticado por las lesiones cutáneas. Actas Dermosifiliogr 2002;93(6):379-83.
- Jesús Duarte-Mote,1 Miguel Jiménez AJ,2 Víctor Lee-Eng C,3 Socorro Romero F,. Relación entre la hemólisis intravascular microangiopática y la disfunción renal en pacientes con preeclampsia. MedIntMex 2013;29:351-355
- A. Cases y G. Escolar. Diátesis hemorrágica en la uremia. NEFROLOGIA. Vol. XVIII. Núm. 4. 1998.
- V. Cararach Ramoneda y F. Botet Mussons Preeclampsia. Eclampsia y síndrome HELLP.2008. Asociación Española de Pediatría.
- Manuel Antonio Díaz de León-Ponce, Jesús Carlos Briones- Microangiopatía trombótica y hemólisis intravascular en hipertensión por embarazo. La mentira del síndrome de HELLP. CirCiruj 2006;74:211-215
- Gonzalo Barrientos, Hernán Michelangelo. Microangiopatia trombotica Medicina (buenos aires) 2006; 66: 289-295.
- Tsai HM. Advances in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. J Am SocNephrol 2003; 14:1072.
- George JN, Vesely SK, Terrell DR. The Oklahoma Thrombotic Thrombocytopenic Purpura-Hemolytic Uremic Syndrome (TTP-HUS) Registry: a community perspective of patients with clinically diagnosed TTP-HUS. SeminHematol 2004; 41:60.
- M. Cuxart, M. Matas, M. Picazo, R. Sans, J. Juvanet y T. Osuna. CASOS CLÍNICOS. Pérdida visual bilateral aguda en pacienteen hemodiálisis. Servicio de Nefrología. Hospital de Figueres. Girona. España.NEFROLOGÍA. Volumen 25. Número 6. 2005.
- Gonzalo Eymin L1, Maricarmen Andrade A2, Max Andresen H3, Jaime Pereira G4 Púrpura trombótico trombocitopénico: Revisión de la literatura a partir de 18 casos.
- Hiroshi Kitamura · Isao Matsui · Noriko Itoh. Tranexamic acid-induced visual impairment in a hemodialysis patient. ClinExpNephrol (2003) 7:311–314.
- M.J. Chueca, S. Berrade, M. Oyarzábal. Talla baja y enfermedades raras. Lowheight and rarediseases.
|
|
|
Discusión |
|
La discusión de este seminario
corresponde al 19 de Septiembre de 2013, a
cargo de Dra. María Cecilia Demaría |
|
| |
|
|
| |
|
|
|
|
|

