|
Discusión del
caso clínico
Dra. Silvina Lema
Se
presenta el caso de una mujer de 17 años de edad con
antecedentes de una glomerulonefritis rápidamente
progresiva (GNRP) a los 10 años que requirió como
tratamiento sustitutivo con hemodiálisis, que terminó
tres años después en un transplante renal de donante
cadavérico. Al ingreso realizaba tratamiento
inmunosupresor con ciclosporina, mofetil-micofenolato y
prednisona. Ingresa a nuestra institución por un cuadro
de tres semanas de lesiones de púrpura palpable en
miembros inferiores hasta glúteos, que remite con el
aumento de los corticoides, falla renal con sedimento
patológico activo, la cual progresa, y en el laboratorio
inmunológico la presencia de ANCA-p positivo (1/640) con
complemento normal. A esto se agrega una nueva biopsia
del injerto que informa como conclusión una
glomerulonefritis asociada a semilunas epiteliales tipo
pauciinmune. Por lo que este cuadro corresponde a
una GNRP en una paciente con trasplante renal.
Ahora
bien, creo que el planteo a partir del cual debe girar
esta discusión es si el cuadro de esta paciente
corresponde a una recidiva de su enfermedad de base que
hace varios años la llevo al trasplante o si estamos
frente a un cuadro de novo.
Comenzaré el
análisis definiendo GNRP: es un síndrome clínico
caracterizado por el desarrollo rápido, a menudo
irreversible, de insuficiencia renal asociado al
desarrollo de lesiones glomerulares inflamatorias con
predominio de proliferación extracapilar y pérdida
progresiva de la función renal en un periodo corto de
tiempo (días, semanas o meses). Su correlato morfológico
clásico es la formación de semilunas en la mayoría de
los glomérulos.
La
severidad de la enfermedad está relacionada en parte con
el grado de formación de semilunas, en los pacientes con
más del 80% de glomérulos con semilunas
circunferenciales tienden progresar su falla renal y no
responder al tratamiento. En comparación, pacientes con
semilunas en menos del 50% de los glomérulos,
particularmente si dichas semilunas son no
circunferenciales, típicamente tienen un curso más
indolente o en raros casos presentan remisión.
El
término GNRP se refiere a glomerulonefritis por
semilunas (crescentic), que es usualmente el
resultado de uno de los 4 desórdenes, los cuales
reflejan diferentes mecanismos de injuria glomerular:
Tipo 1: Anti-membrana basal gromerular (MBG):
Enfermedad por anticuerpos anti-MBG. (enfermedad
anti-MBG, síndrome de Goodpasture)
Tipo 2: Por inmunocomplejos:
puede: 1) ser idiopático, 2) representar una reacción a
un estímulo antigénico conocido (por ejemplo,
glomerulonefritis desencadenada por endocarditis
bacteriana o infección estreptocócica, o infección por
virus de las hepatitis B o C en caso de
glomerulonefrítis crioglobulinémicas). o bien 3) formar
parte de un trastorno multigeneralizado por complejos
inmunitarios (ej.
Nefritis lúpica, Nefropatía por IgA).
Tipo 3: Pauci-inmune:
Hay una glomerulonefritis necrosante pero pocos o ningún
depósito por inmunofluorescenca o microscopia
electrónica. La mayoría de los pacientes con vasculitis
limitada al riñón son ANCA positio, con 75 a 80% de
mieloperoxidasa (MPO)-ANCA, y muchos tienene o
desarrollarán síntomas sistémicos de una vasculitis.
Los pacientes con ANCA negativos son considerados parte
de este espectro, y a menudo tienen clínica, hallazgos
de biopsia y pronóstico similares.
En este grupo se incluye a la
granulomatosis de Wegener, poliangeítis microscópica y
vasculitis limitada al riñón que eventualmente puede
progresar a vasculitis sistémica.
Por lo dicho anteriormente puede considerarse “Tipo ANCA
+”.
Tipo 4: Enfermedad con anticuerpos dobles positivo:
ANCA-Anti MBG.
Tipo 5: Idiopática:
Este término es aplicado a dos casos: enfermedad por
inmnocomplejos (tipo 2) que no clasifica para ninguna
categoria identificable, y una pauci-inmune (tipo 3) que
es ANCA negativo.
Para
comenzar tengo que hacer un breve comentario de la forma
de presentación del cuadro actual. De acuerdo a los
datos que se obtuvieron en un primer momento, uno de los
diagnóstico que se planteó fue el de Púrpura de
Schönlein-Henoch. El mismo es un cuadro de
vasculitis generalizada singular que se caracteriza por
la presencia de púrpura palpable (distribuida
principalmente en las nalgas y los miembros inferiores),
artralgias, signos y síntomas gastrointestinales y
glomerulonefritis. Es una vasculitis de los vasos de
pequeño calibre. Predomina en niños entre dos y diez
años de edad, aunque puede presentarse luego de los 15
años. Su etiología es desconocida, es considerada un
subgrupo de vasculitis por hipersensibilidad pero no se
ha identificado el alérgeno específico. Como la lesión
glomerular es idéntica a la observada en caso de
nefropatía por IgA, la nefritis de Henoch-Schönlein y la
nefropatía por IgA pueden ser parte de un espectro de
manifestaciones de una sola enfermedad. El díagnóstico
de púrpura de Henoch-Schönlein se basa en los signos y
síntomas clínicos. La biopsia cutánea es de utilidad
para confirmar una vasculitis leucocitoclástica
con depósito de IgA y C3 en el estudio con
inmunofluorescencia.
Rara vez se
necesita biopsia renal para el diagnóstico, cuando se
realiza muestra los mismos depósitos de IgA y fibrina en
el mesangio. Se han comprobado recidivas de la
nefropatía, especialmente por IgA, en los pacientes
trasplantados.
En
nuestra paciente tenemos una forma de presentación
característica, la edad de la misma y la notable mejoria
de las lesiones en piel con el aumento de los
corticoides. Podríamos considerarla como una enfermedad
de novo ya que la primera biopsia renal no mostraba los
depósitos, sin embargo en la nueva biopsia del injerto
tampoco los presenta por lo que este diagnóstico
realizado antes de su internación queda descartado.
Con
el laboratorio inmunológico actual donde informa la
presencia de ANCA p positivo por inmunofluorescencia (IF),
con especificidad por EIA por mieloperoxidasa a títulos
altos y una biopsia del injerto que informa
glomerulonefritis asociada a semilunas epiteliales sin
evidencia de depósitos por inmunofluorescencia, tenemos
el diagnóstico de una GNRP pauci-inmune o asociada
con ANCA.
Las
principales glomerulonefritis pauciinmunitarias son
glomerulonefritis idiopática con semilunas limitada al
riñón, poliarteritis nudosa microscópica y
granulomatosis de Wegener (GW) .La enfermedad
glomerular significativa puede complicar también el
síndrome de Churg-Strauss y las formas clásicas de
poliarteritis nudosa (PAN), aunque con menos frecuencia
que a los tres trastornos que se acaban de mencionar.
La
notable superposición de manifestaciones clínicas y la
histopatología glomerular, y la presencia de ANCA
circulantes en la mayoría de los pacientes, sugieren que
estas entidades constituyen un espectro de la misma
enfermedad. En realidad, las enfermedades de este grupo
se clasifican frecuentemente bajo el término global
vasculitis de vasos pequeños relacionada con ANCA.
En la
GW ocurre lesión renal en 80% de los pacientes, y
varía entre inflamación indolente pero sostenida e
insuficiencia renal rápidamente progresiva. Se
identifican ANCA citoplásmicos en el momento de la
presentación, también en 80% de los pacientes que tienen
enfermedad renal, y en 10% más durante el seguimiento.
La biopsia renal revela de manera característica
glomerulonefritis pauciinmunitaria focal necrosante con
formación de semilunas. En contraste con lo que ocurre
en los pulmones, es raro que se encuentren granulomas en
los riñones.
Esencialmente todos los pacientes con GW clásica o
limitada presentan afectación de la vía aérea superior o
pulmón, y en la mayoría de ambos. Aproximadamente el 90
% de los pacientes con enfermedad activa generalizada
tienen ANCA positivo, por lo que claramente hay un
porcentaje con enfermedad activa y ANCA negativo. Además
en formas limitadas de la enfermedad (con síntomas
predominantemente de vía aérea y ausencia de afectación
renal), más del 40% son negativos. Por lo que la
ausencia de ANCA no excluye el diagnóstico de GW. En
grandes estudios la sensibilidad de ANCA-PR3 para GW
está en relación con la extensión, severidad y actividad
de la enfermedad. Entre los pacientes con GW con ANCA,
el 80 a 90% tienen PR3-ANCA. El resto tiene MPO. El
diagnóstico es sugerido por la clínica, los hallazgos de
laboratorios, la presencia de ANCA y debería ser
confirmado por biopsia tisular del lugar de actividad de
la enfermedad.
La
poliangeítis microscópica (PAM) es una enfermedad
diseminada que se caracteriza por vasculitis necrosante
que abarca múltiples aparatos y sistemas como pulmones,
piel, articulaciones y riñones. Esta enfermedad suele
acompañarse de glomerulonefritis y capilaritis pulmonar.
Se dice que la ausencia de inflamación granulomatosa en
la poliangitis microscópica la distingue de la
granulomatosis de Wegener. Se identifican ANCA
circulantes en 70% de los pacientes. En contraste con la
GW, la mayoría son ANCA-MPO, con una minoría que
presenta PR3. Como PR3-ANCA o MPO-ANCA podrían estar
presentes en ambas enfermedades, GW y PAM, estas
enfermedades no pueden ser distinguidas en base a la
especificidad del ANCA. Además, la falta de consensos
para definir la poliangeítis microscópica complican la
interpretación de los datos de laboratorio. Por su
predilección por los vasos pequeños, la poliangitis
microscópica y la granulomatosis de Wegener comparten un
cuadro clínico similar.
En
1982, fueron descriptos por primera vez los ANCA
(anticuerpos contra citoplasma de neutrófilos), en
pacientes con glomerulonefritis pauci-inmune. Sin
embargo en 1985 fueron asociados con GW. Con el correr
de los años se fue estableciendo la relación entre los
ANCA, la granulomatosis de Wegener, la poliangeítis
microscópica y la glomerulonefritis idiopática con
semilunas limitada al riñón .En estas condiciones
los ANCA tienen especificidad por alguno de los
antígeno, PR3 o enzima MPO, pero nunca por ambos. En
contraste con estas condiciones, la poliarteritis
nodosa no está asociada a ANCA por lo que se puede
descartar si el mismo es positivo.
Si analizamos
los antecedentes, esta paciente presentó una GNRP con un
laboratorio inmunológico con ANCA negativo. Por lo que
me pregunto si este dato de laboratorio me descarta el
diagnóstico de pauci-inmune en el primer episodio de
falla renal. De acuerdo a lo estudiado, como el 40% de
los pacientes con GW limitada y 10% con enfermedad grave
no tienen ANCA, un laboratorio negativo no lo excluye.
Lo mismo para poliangeítis microscópica, donde 30% son
ANCA (-). Además el estatus de ANCA podría cambiar con
el tiempo, un paciente que es ANCA negativo en su
presentación con síntomas constitucionales e infiltrados
pulmonares podría transformarse en positivo para
PR3-ANCA al desarrollar una enfermedad más generalizada,
por ejemplo al agregar una glomerulonefritis.
Una
pregunta que me hago es si la medición de los títulos de
ANCA pueden predecir la recidiva de la enfermedad.
En un
estudio realizado entre 1996 y 1998, se tomaron 100
pacientes con granulomatosis de Wegener y con ANCA
positivo, y se hicieron titulaciones cada dos meses de
dichos anticuerpos. Mientras se los evaluó clínicamente
para recaída de la enfermedad. El grupo de médicos que
evaluaba recaída, desconocía las titulaciones. Los
resultados fueron: 77 % tuvieron aumento de ANCA, de los
cuales 43% no presentaron recaídas, 44% si tuvo recaída
y hubo un valor predictivo positivo entre 57 y 100% de
acuerdo a la técnica utilizada, ya sea IF o ELISA y de
acuerdo a la especificidad del ANCA. La conclusión a la
que arribaron fue que las mediciones seriadas de ANCA
son útiles para predicción de recaída en GW. Sin embargo
si observamos, existe un porcentaje casi idéntico de
pacientes que no tuvieron recaída, por lo que esa
conclusión no me parece adecuada.
Además existen reportes como por ejemplo el caso de una
paciente con similares características a la nuestra: (12
años) con GNRP con MPO-ANCA (+), con diagnóstico de
poliangeítis microscópica que realizó en un primer
momento tratamiento inmunosupresor, a los 6 años
requirió hemodiálisis y 9 meses después se realizó
trasplante renal. En su seguimiento no presentó
recaídas, como lo muestra el siguiente gráfico:
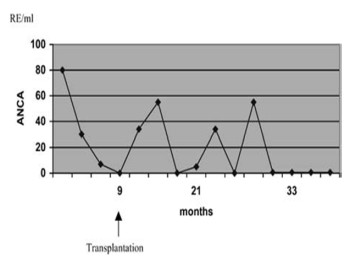
Niveles de ANCA después de entrar en enfermedad renal
etapa terminal. El punto 0 es el ingreso a hemodiálisis.
En el
caso de nuestra paciente que es una trasplantada renal,
¿qué sucede? ¿puede haber recaída en el injerto?
En
realidad, en este tema existe poca experiencia. Reportes
aislados indican recidiva con manifestaciones renales y
extrarrenales en trasplantados. Sin embargo se opina que
el riesgo podría ser menor en trasplantados que en
pacientes en diálisis por la inmunosupresión de los
primeros para prevención de rechazo del injerto. Las
recaídas, particularmente si son severas, usualmente
requieren ciclofosfamida y suspender otras drogas.
Con
respecto al pronóstico de supervivencia, en un estudio
del 2007, los resultados de 19 trasplantados con GW o
PAM fueron comparados con 38 controles sin vasculitis.
Ambos grupos tuvieron similar supervivencia actuorial a
10 años (87 y 90% respectivamente).
Para
concluir, nuestra paciente tiene una recidiva de su
enfermedad de base que es una vasculitis asociada a
ANCA. Y por las características clínicas, la biopsia
renal y la presencia de ANCA MPO a titulaciones altas,
hace diagnóstico de poliangeítis microscópica.
Con
respecto a las conductas a seguir creo que esta paciente
debe tener un seguimiento estricto tanto clínico como de
laboratorio ya que por su severa inmunodepresión tiene
alto riesgo de infección y por la presencia de síntomas
de vías respiratorias altas podría complicarse con
afectación pulmonar, cuadro de alta mortalidad.
Y
dejo un interrogante, si eventualmente no respondiera al
tratamiento, y considerando que estamos frente a una
enfermedad sistémica recidivante, estaría indicado un
nuevo transplante renal.
BIBLIOGRAFIA:
1.
Boomsma MM; Stegeman CA; van der Leij MJ; Oost W;
Hermans J; Kallenberg CG; Limburg PC; Tervaert JW. “Prediction
of relapses in Wegener's granulomatosis by measurement
of antineutrophil cytoplasmic antibody levels: a
prospective study”.
Arthritis Rheum 2000 Sep;43(9):2025-33.
2.
Moroni G; Torri A; Gallelli B; Quaglini S; Pozzi
C; Banfi G; Poli F; Montagnino G; Ponticelli C; Messa P.
“The long-term prognosis of renal transplant in
patients with systemic vasculitis” Am J Transplant.
2007 Sep;7(9):2133-9. Epub 2007 Jul 19.
3.
Gera M; Griffin MD; Specks U; Leung N; Stegall
MD; Fervenza FC. “Recurrence of ANCA-associated
vasculitis following renal transplantation in the modern
era of immunosupression”. Kidney Int. 2007
Jun;71(12):1296-301. Epub 2007 Apr 4.
4.
“Successful renal transplantation in a child with
ANCA-associated microscopic polyangiitis”.
Pediatr Nephrol (2003) 18:696–699.
5.
Principios de Medicina interna. Harrison, 16ta
edición pagina 1809 a 1864 y 2211 a 2213
6.
Medicina Interna. Farreras-Rozman 11 edición
pagina 908 a 961
|

