|
Discusión del
caso clínico
Dr. Francisco Consiglio
Se discute el caso de un varón de 58 años que ingresa por 1 mes de pérdida de peso, astenia y que agrega de una semana signos de sangrado a nivel cutáneo, evidenciándose al examen además poliadenopatías, y al laboratorio pancitopenia.
Como datos de jerarquía:
- Pancitopenia con plaquetopenia severa.
- Complemento consumido: CH50: 20 (24-56) C3: 91 (103-145) C4: 19 (20-50)
- FAN, Anti-ADN, Anti-Ro positivo.
- ANCA P (1/160).
- HIV, VHC, VHB negativos. CMV, VEB IgG positivo, IgM negativos.
- Médula ósea: Levemente hipocelular (celularidad global 40%) con predominio de la serie roja, disminución moderada de la serie granulocítica y severa hipoplasia de la progenie megacariocítica.
- Sin respuesta al uso de corticoides e Hidroxicloroquina.
- Desarrolla shock de probable origen séptico, con foco cutáneo.
- Procalcitonina 2,1 ng/ml.
- Hemorragia pulmonar.
- Se indica inmunoglobulina endovenosa, sin obtener respuesta.
- Inicia ciclofosfamida, posteriormente síndrome de falla multiorgánica y fallece.
Datos guía para el desarrollo de la discusión:
- Pérdida de peso, astenia, poliadenopatías y pancitopenia, con plaquetopenia severa.
- FAN, Anti ADN nativo, Anti Ro positivos. Complemento consumido.
- Médula ósea levemente hipocelular, con severa hipoplasia megacariocítica.
- Desarrollo de hemorragia pulmonar.
Interrogantes y objetivos de la discusión.
- ¿Se trata de un paciente con Lupus Eritematoso Sistémico?
- ¿Puede existir una causa farmacológica subyacente?
- ¿Cómo interpretar la trombocitopenia?
- ¿Cómo interpretar la hemorragia pulmonar?
- Valor del ANCA y procalcitonina en LES.
- Conclusión.
En cuanto al primer interrogante, ¿Tiene este paciente un lupus eritematoso sistémico (LES)?
El lupus eritematoso sistémico es una enfermedad de causa desconocida que puede afectar a cualquier aparato o sistema del organismo, y ocasionar una gran variedad de manifestaciones clínicas. Si bien tiene predilección sobre el sexo femenino, esto es más marcado en los adultos jóvenes (9/1), siendo en mayores de 65 años la incidencia 2/1. Se trata de una enfermedad crónica, que cursa en brotes, para la cual no existe aún tratamiento curativo, sin embargo su pronóstico ha mejorado en relación al diagnóstico precoz y las nuevas pautas terapéuticas.
En cuanto a la sintomatología general el paciente presenta astenia, malestar general y pérdida de peso. Hasta la mitad de los casos puede presentar adenopatías, sobretodo en fase de actividad de la enfermedad, y han resultado al examen anatomopatológico compatibles, descartándose razonablemente otras posibles causas que las expliquen. Presenta además serositis, como es más frecuente en hombres, evidenciada por derrame pleural izquierdo. A nivel hematológico trombocitopenia, leucopenia y linfopenia. El laboratorio inmunológico informa anticuerpos antinucleares a títulos muy elevados (1/5120), y marcadores serológicos positivos Anti ADN (Alta sensibilidad y especificidad- 70%, 95%). Anti Ro (Baja sensibilidad, Alta especificidad 25 a 24%; 87 a 95%) (1).
Estrictamente, de los criterios de clasificación propuestos por la American College of Rheumatology presenta:
- Trastornos hematológicos: plaquetopenia-linfopenia.
- Serositis: derrame pleural unilateral izquierdo.
- Factor antinuclear positivo, y a títulos muy elevados.
- Marcadores serológicos: Anti ADN nativo positivo.
Según los criterios propuestos, 4 o más de manera simultánea, o secuencial presentan una sensibilidad y especificidad para el diagnóstico del 96%. (2,3)
Considero en este caso entonces el diagnóstico de LES muy probable.
Surge el interrogante acerca de la posibilidad de seudolupus secundario a drogas, puntualmente por fenitoína. En este sentido se conoce que numerosas drogas inducen un cuadro clínico e inmunológico prácticamente idéntico al LES (Tabla 1), aunque más frecuentemente solo se detecte en el laboratorio FAN positivo. La sintomatología, aunque muy parecida tiene algunas diferencias con el LES idiopático. El proceso es reversible tras la retirada del medicamento, las manifestaciones (Tabla 2) incluyen síntomas generales, artralgias, artritis, serositis, mientras que la afección neurológica y renal es rara. En cuanto al perfil inmunológico el FAN es positivo en el 95% de los casos, el anti ADN nativo suele ser negativo, y en el 90% de los casos los anticuerpos anti-histonas son positivos, el complemento suele ser normal, y los ENAS también suelen ser negativos.
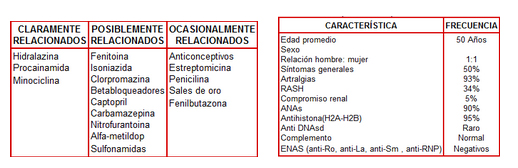
Tabla 1: Drogas relacionadas al desarrollo de Lupus
Tabla 2: Características diagnósticas de lupus secundario a drogas.
El cuadro que presenta el caso en discusión no presenta las características clínicas, ni de laboratorio compatibles con lupus secundario a drogas, por lo que considero esta hipótesis alejada. (1)
Para el análisis de la trombocitopenia es conveniente comenzar con aspectos generales. Se entiende por trombocitopenia valores de plaquetas en sangre periférica < 150.000/microL. Las causas que la puedan desencadenar, de acuerdo al mecanismo fisiopatogénico son: a) Por disminución en la producción, b) Por incremento de la destrucción, c) Dilucional. D) Distributiva. (4).
a) Por disminución de la producción plaquetaria.
- Luego de infecciones virales. (Rubeola, parotiditis, varicela, parvovirus, VHC y virus Epstein-Barr).
- HIV.
- Postquimioterapia, o menos frecuentemente radioterapia.
- Aplasia, o hipoplasia, congénitas o adquiridas.
- Toxicidad por alcohol.
- Déficit vitamina B y ácido fólico.
b) Por destrucción periférica:
-Fármacos: heparina, quininas, quinidinas, ácido valproico.
-Destrucción aloinmune: posttranfusional, neonatal, post transplante.
-Coagulación intravascular diseminada.
-Púrpura trombótica trombocitopénica.
-Síndrome HELLP.
-Infecciosas: Mononucleosis, citomegalovirus.
-Misceláneas: Bypass cardiovascular, aneurimas aórticos muy extensos, lesiones intravasculares, o metastásicas intracardíacas.
-Inmunes: Trombocitopenia inmune idiopática (TIP), trombocitopenia asociada al lupus eritematoso sistémico, y otras trombocitopenias inmunes secundarias más raras (SAF, Inmunodeficiencia variable común, Leucemia -linfática crónica, Síndrome de Evan´s, Síndrome autoinmune linfoproliferativo, HIV, VHC, H. pylori, postvaccinal, misceláneas).
c) Dilucional: En el contexto de transfusiones de unidades de glóbulos rojos masivas.
d) Distributiva: En pacientes con esplenomegalia, en hipertensión portal, por secuestro esplénico.
Las causas dilucional y distributiva se encuentran alejadas, puesto que el paciente no ha recibido transfusiones masivas de glóbulos rojos, ni presenta esplenomegalia, que genere secuestro de plaquetas, como se suele observar en pacientes con esplenomegalia congestiva por hipertensión portal.
En cuanto a las causas de destrucción periférica, por anamnesis, examen físico y estudios complementarios la mayoría quedan descartadas. En cuanto a las causas inmunes se aleja la TIP como etiología del cuadro actual, pues existen en la historia clínica datos sugestivos de trombocitopenia inmune secundaria, y además la hipoplasia de la serie megacariocítica no es lo habitual.
El hallazgo de una médula ósea normocelular (40%) con hipoplasia marcada de la serie megacariocítica nos orienta hacia la denominada púrpura trombocitopénica amegacariocítica. Se trata de un trastorno hematológico inusual, que se caracteriza por trombocitopenia, usualmente <20.000/mm3. asociada a desde una marcada disminución hasta ausencia total de megacariocitos en médula ósea, con el resto de las series conservadas (5,6).
Existen causas congénitas y adquiridas. La forma congénita suele presentarse en la infancia, a edades muy tempranas, por lo que se aleja del caso en discusión. En esta forma estarían implicadas mutaciones del gen que codifica c-mpl, el receptor de la trombopoyetina (TPO). Estos pacientes suelen responder a la terapia recombinante con IL-3, aunque el tratamiento de elección parece ser el transplante de médula ósea.
Las formas adquiridas se suelen presentar clínicamente de manera indistinguible de los pacientes con trombocitopenia inmune, con disminución marcada de las plaquetas solamente, y con escasos signos de sangrado, aun con valores extremadamente bajos de plaquetas. Se postula que este fenómeno se debe a que las plaquetas jóvenes son más activas desde el punto de vista hemostático, y así no ocurren hemorragias severas con valores peligrosos se plaquetas. El factor que logra diferenciar estas entidades, la trombocitopenia inmune primaria de la amegacariocítica, es el examen de médula ósea, que muestra en esta última una disminución marcada hasta la abolición de la serie megacariocítica.
Hay varias causas implicadas en esta entidad, entre ellas tóxicas, drogas, infecciones virales, enfermedades sistémicas como el lupus, y déficits de citoquinas. En cuanto a las causas tóxicas el benzeno, oro, mercurio, e insecticidas están implicados. El paciente no presenta antecedente de exposición a estos tóxicos. En cuantos a causas farmacológicas se encuentran alejadas por la falta de su uso, están descriptas la clorotiazidas, estrógenos, el etanol, y el cloranfenicol. Infecciones virales por citomegalovirus (CMV), o parvovirus B-19 (PB-19) también están descriptas, contamos con IgM negativa para CMV, y no se ha estudiado PB-19, por lo que no se puede descartar, aunque existe otra causa más probable. Tampoco se han realizado dosajes de citoquinas.
La trombocitopenia inmune secundaria a LES, ocurre en un 7 a 30% de los pacientes con LES en las diferentes series, y constituye marcador pronóstico seguro, que identifica pacientes que cursan actividad lúpica actual, en los que se está produciendo daño orgánico por la enfermedad, constituyendo un signo de mal pronóstico.
La fisiopatogenia es multifactorial, e incluye mecanismos asociado a anticuerpos dirigidos contra varios blancos, el principal es contra las glicoproteínas de las plaquetas, GP IIb/IIIa y GPIb/IX (como en la PTI), que ligadas a las plaquetas, son fagocitadas por los macrófagos en el bazo. Anticuerpos contra la TPO y c-mpl han sido también reportados. A su vez se decriben anticuerpos contra el ADN, los fosfolípidos y las proteínas ligadas a fosfolípidos, el ligando CD-40L, e inmunocomplejos de diversa composición, que generan apoptosis al afectar al FAS-FAS ligando. A su vez la trombocitopenia puede producirse en el contexto de exacerbación de la enfermedad por vasculitis sistémica, síndrome hemofagocítico, en el contexto de microangiopatía trombótica, amegacariocitosis, y por daño medular por fármacos, los más comunes son AINES, estatinas, inhibidores de la bomba de protones, antibióticos, e inmunosupresores como al azatioprina, la ciclofosfamida, y el metotrexate (7,8,9).
En cuanto al mecanismo que esté mediando la enfermedad en el caso en discusión, es muy probable que se trate de afección de la trombopoyesis, debido a la disminución marcada de los megacariocitos en la médula ósea, es decir estemos frente a una trombocitopenia amegacariocítica adquirida (AATP) asociada a LES. En este sentido existen estudios que han demostrado mecanismos fisiopatogénicos en pacientes con LES. Kuwana et al. (10) ha reportado la presencia de anticuerpos contra el receptor de trombopoyetina (Anti c-mpl) en pacientes con LES trombocitopénicos, y que esto se asocia a disminución de la serie megacariocítica en médula ósea. A su vez, en este grupo de pacientes ha constatado niveles significativamente elevados de TPO, y postula una interesante hipótesis, que los anticuerpos anti c-mpl bloqueen la unión de la TPO a sus receptores, afectando así la trombopoyesis en paciente con LES.
En Rheumatology 2006 el mismo grupo de investigadores publica un estudio que demuestra que el perfil de anticuerpos que presentan los pacientes lúpicos trombocitopénicos se asocian a diferentes fenotipos de trombocitopenia, en términos de densidad de megacariocitos en médula ósea y respuesta terapéutica a los regímenes estándar de trombocitopenias inmunes. Así aquellos pacientes con anticuerpos anti GPIIb/IIIa únicamente presentaban una médula ósea normal, o hiperplasia megacariocítica. En cambio aquellos Anti-TPOR positivos se asociaron de manera significativa a hipoplasia megacariocítica, y pobre respuesta a corticoides e inmunoglobulinas. De esta manera jerarquiza la importancia del dosaje de estos autoanticuerpos, y su utilidad para predecir el fenotipo de trombocitopenia, y su respuesta terapéutica en lúpicos (11).
En cuanto al tratamiento se han reportado diferentes esquemas terapéuticos en AATP asociada a LES, y hay que aclarar que no existe un tratamiento estandarizado como en otras entidades como la TPI, y que la evidencia que sustenta su uso es pobre, de reportes o pequeñas series de casos.
Los corticoides han sido utilizados, desde el primer reporte de AATP asociada al LES, en 1970, con resultados poco prometedores, a manera de pulsos de metilprednisolona, seguidos de prednisona a dosis inmunosupresoras, extrapolando quizás la terapia estándar utilizada en ITP, grupo en el cual tienen una muy buena respuesta, con el uso de dexametasona en ciclos del 80-90% a los 5-7 días, con un 40-70% de respuesta sostenida. Incluso en un reporte reciente se ha visto la mala respuesta asociada al uso de corticoides en pacientes Anti c-mpl positivos, que se asocia al fenotipo megacariocítico hipoplásico en médula ósea. Con el uso de inmunoglobulinas de igual manera se ha visto una pobre respuesta, dado que la acción principal de la IgIV se cree que es la interacción entre la porción Fc de la infusión de inmunoglobulinas y los receptores Fc en las células diana, es probable que la inmunoglobulina intravenosa tenga poco efecto sobre el bloqueo de la señal TPO a través de la región variable de los anticuerpos (11,12).
El uso de ciclosporina A ha sido reportado en múltiples oportunidades en AATP, con resultados favorables, e incluso en trombocitopenia asociada a LES ha demostrado ser efectiva. El principal problema es su latencia hasta obtener respuesta terapéutica, varias semanas a meses, y efectos adversos como la hipertensión y la nefrotoxicidad han limitado su uso (13,14).
La remisión completa con globulina anti-timocito ha sido reportada, también en este caso el principal problema es la prolongada latencia hasta el efecto terapéutico deseado (15).
El uso del Rituximab, un anticuerpo monoclonal que causa depleción de las células B, a través de su unión a CD-20 en pacientes con AATP se ha descripto, existiendo un reporte incluso en AATP asociado a LES, en una mujer de 42 años, que no presentaba respuesta a prednisona y ciclosporina A, y que con esta droga mejora drásticamente su recuento de plaquetas, además observaron que los niveles de anti c-mpl descendieron de manera paralela, sugiriendo que el rituximab logra eliminar la producción de estos anticuerpos implicados en la fisiopatogenia de la trombocitopenia amegacariocítica. Sin embargo, entre sus efectos adversos, el riego de infecciones severas está descripto, en 2006 la FDA advierte de dos casos de pacientes con LES que fallecen por leucoencefalopatía multifocal progresiva asociada a reactivación del virus JC (16).
También existe en la literatura un único reporte de uso de eltrombopag, un agonista del receptor de la trombopoyetina, en un paciente con AATP y LES, con resultado favorable. Se trata de una molécula no peptídica mimética a la TPO que se liga a un sitio diferente al de la TPO en el receptor c-mpl, estimulando la producción plaquetaria a través de la fosforilación de JAK2/STAT5 y MAPK. Esta droga se administra por vía oral, y se ha visto que su ejerce una acción dosis dependiente comenzando alrededor de los 8 días, con un pico a los 16 días. En un estudio doble ciego, ramdomizado y controlado, que evaluaba la eficacia de esta droga a 3 dosis diferentes en pacientes con ITP, considerando el endpoint primario plaquetas >50.000/microL. lograron en un 28%, 79%, y 81% este objetivo con 30, 50, y 75 mg respectivamente. Otro dato de jerarquía que se desprende de este trabajo es que al día 15 de tratamiento más del 80% de los pacientes que recibían 50 o 75 mg de eltrombopag/día presentaban plaquetas >50.000microL. La incidencia y severidad de los efectos adversos fue similar que en el grupo placebo. Lo más frecuente es la cefalea (17%).
En este reporte se propone a los agonistas de TPOR como una alternativa válida, cuando en pacientes con AATP en general fallan los corticoides orientada inicialmente la terapéutica como ITP, ya que brinda eficacia, un perfil seguro y como principal ventaja puede reemplazar la estrategia inmunosupresora (17).
Otros tratamientos que se pueden utilizar en el manejo de la trombocitopenia en LES en general, dejando de lado el mecanismo subyacente (18), son:
-
La esplenectomía, constituye una opción de tratamiento que ha sido eficaz en una serie reciente en 64% de pacientes, con una respuesta sostenida, a lo largo de más de 6 años de seguimiento, sin embargo la mitad de los mismos requirió terapia médica adyuvante. De los pacientes que sufrieron recaídas la mitad respondió posteriormente al tratamiento médico. Se reserva su uso para pacientes que no responden a los tratamientos convencionales.
- La azatioprina ha sido utilizada para el tratamiento de anemia hemolítica, y trombocitopenia inmune a pesar de la falta de estudios clínicos randomizados. Sin embargo, puede causar toxicidad medular en pacientes con déficit de actividad de la tiopurina-metiltransferasa. Por este motivo se recomienda su dosaje previo al inicio de esta droga en pacientes lúpicos con citopenias.
-
El mofetil-micofenolato (MMF) es otro agente ahorrador de corticoides que ha sido utilizado para el tratamiento de pacientes lúpicos con citopenias refractarias. Se han reportado casos de anemia hemolítica, trombocitopenia, y aplasia de células rojas asociados a LES que han respondido a MMF. A pesar de no existir estudios prospectivos que demuestren su eficacia, su uso se está incrementando en manifestaciones no renales en lupus.
- La ciclofosfamida (CYC) es un agente alquilante comúnmente utilizado en la inducción de remisión de la nefritis lúpica. Un estudio retrospectivo describe 7 pacientes con trombocitopenia resistente a los corticoides que responden tras 1 a 4 dosis de CYC. También se ha utilizado en series de casos y reportes aislados en TTP, y anemia aplásica. El principal peligro, como en el caso de la azatioprina es la toxicidad medular.
-
El transplante de médula ósea ha sido investigado en LES, y se ha reportado un 44% de pacientes libres de enfermedad a los 5 años, con una sobrevida global del 76%. Se prefiere el transplante autólogo a los alogénicos, debido al menor riesgo de toxicidad severa. Sin embargo, dado la relativa alta tasa de mortalidad asociada al procedimiento, actualmente se limita su uso solo para pacientes refractarios a los tratamientos convencionales.
En cuanto al desarrollo de hemorragia pulmonar, esta se hace evidente por presentarse con la triada clásica, hemoptisis, nuevos infiltrados en la radiografía de tórax, y caída de la hemoglobina. Si bien existen múltiples causas de hemorragia pulmonar, en el contexto de este paciente con diagnóstico muy probable de LES, y en actividad franca, con plaquetopenia severa, es lógico atribuírselo al mismo origen. Es difícil diferenciar en este caso si se trata de un proceso inmune con alveolitis, o constituye un sangrado mayor asociado a plaquetopenia severa, o ambos, secundarios a LES. A favor de diátesis hemorrágica por plaquetopenia en la evolución presenta además sangrados por sitios de venopunción y mucosos.
En cuanto a hemorragia alveolar difusa en lúpicos presenta una incidencia entre 2 y 5.4%, con una mortalidad elevada del 23 al 92% (19). La afectación renal tiene una relación estrecha con la manifestación clínica del síndrome de hemorragia alveolar (93%), generalmente como una condición preexistente, que no está presente en este caso.
En la tercera parte de los casos los pacientes padecen una infección viral o bacteriana aguda, sobre todo los que cursan tratamiento inmunosupresor. En estos pacientes es prudente descartar infecciones virales por citomegalovirus, herpes virus o bacterianas como Legionella y estafilococo (20,21).
La capilaritis se informa en las series hasta en 80%, aunque no es específica de LES. Los estudios de inmunofluorescencia de tejido obtenido por biopsia pulmonar abierta o transbronquial prueban en la mayoría de los pacientes con LES, el depósito de complejos inmunitarios, especialmente IgG y otros anticuerpos, así como depósitos de C3 dentro de las paredes alveolares, el intersticio y las células endoteliales. Existen muchas similitudes entre la neumonitis lúpica y la hemorragia alveolar en pacientes con LES, por lo que se ha propuesto que ambas enfermedades tienen la misma forma clínica que se refleja en diferentes espectros, caracterizadas ambas por lesión a la unidad alvéolo-capilar.
En cuanto al tratamiento consiste en corticoides, especialmente bolos de metilprednisolona, y ciclofosfamida. Se ha reportado también el uso de plasmaféresis en esta entidad.
Otras causas de hemorragia pulmonar se alejan del caso en discusión, como el síndrome de Goodpasture, la granulomatosis de Wegener, o la Poliangeítis microscópica, de todas maneras es importante la solicitud de serologías, puesto que resultados positivos en este sentido nos obligarían a plantear otras conductas terapéuticas.
Probablemente en el desarrollo de la hemorragia pulmonar en este caso estén interpuestos varios mecanismos, autoinmune, por plaquetopenia, alteraciones en la coagulación por sepsis, e infeccioso en el contexto shock de probable etiología séptica, en un paciente inmunosuprimido. Lamentablemente el estado del paciente no permitió la realización de una broncofibroscopía que nos hubiese dado información muy valiosa para el diagnóstico.
Algunos datos de los estudios complementarios que merecen un análisis:
En cuanto al valor de los ANCA en el escenario del paciente lúpico cabe destacar que hay numerosas publicaciones con resultados controvertidos (22,23,24). En un estudio realizado por el Cervera y colaboradores, concluye que la prevalencia de ANCA asociada a LES es del 16,4%, más frecuentemente se constatan ANCA-p. Si bien estarían asociados a serositis, artritis, livedo reticularis y trombosis venosa, no es posible otorgarles un rol fisiopatológico en estos eventos. Actualmente sabemos que representan marcadores de actividad sin ejercer un rol patogénico, si bien tampoco puede descartarse que representen sólo un epifenómeno de autoinmunidad.
Por último algunas consideraciones sobre la procalcitonina. Es sumamente importante distinguir intercurrencia infecciosa y reactivación en pacientes con LES con síndrome febril, ya que conllevan diferente pronóstico, terapéutica y seguimiento.
La PCT incrementa su valor ante infecciones bacterianas severas, fúngicas y parasitarias, manteniéndose normal en las reactivaciones, las infecciones virales o por micobacterias. Cabe destacar que no se ve influenciada por la insuficiencia renal ni por la terapia inmunosupresora, escenario frecuente en estos pacientes. Tiene una baja sensibilidad (65%), con un valor predictivo positivo del 89% utilizando como punto de corte un valor de 0,5 ng/ml y 100% usando un valor de 1,2 ng/ml (25,26, 27). Es importante destacar que los biomarcadores deben ser útiles como herramientas para el razonamiento clínico, pero no deben ser nuestra única guía al momento de elaborar un diagnóstico, plantear un plan terapéutico, o realizar el seguimiento en un paciente.
Como conclusión estamos frente a un paciente con diagnóstico muy probable de lupus eritematoso sistémico, que como principal manifestación muestra trombocitopenia, y que teniendo en cuenta los hallazgos en la anatomía patológica de la médula ósea se lo puede considerar una trombocitopenia amegacariocítica adquirida, asociada a LES. Como hemos visto en algunos estudios es probable que este caso se incluya en el fenotipo de lúpicos con trombocitopenia y producción de Anti TPOR, que se asocia a hipoplasia severa megacariocítica, y que presenta una mala respuesta al tratamiento con corticoides e inmunoglobulinas.
En relación a la hemorragia alveolar no contamos con broncofibroscopía, por lo tanto no se puede establecer una causa responsable. Es probable que responda a una etiología multifactorial.
En cuanto a la terapéutica, como hemos visto agentes como la ciclosporina A, o la globulina anti-timocito han demostrado ser efectivos en AATP, pero presentan un prolongado período de latencia, por lo que su uso en este caso no sería adecuado.
En cuanto al rituximab ha sido utilizado con éxito en AATP en LES, planteándose como principal mecanismo al depletarse las células B, la disminución en la producción de anti TPOR, y así la normalización del recuento de plaquetas. Su principal problema es el desarrollo de infecciones severas.
Por último el uso de eltrombopag, ha sido reportado en un caso, y sería una opción más que interesante, dado su mecanismo de acción que tiene como diana al receptor c-mpl, llave de la producción plaquetaria.
Considerando el crítico estado del paciente, y que cursaba tratamiento con antibióticos de amplio espectro, ante la posibilidad de alveolitis y como tratamiento para la trombocitopenia, creo oportuno haber iniciado CYC, el principal temor en este sentido a corto plazo es el empeoramiento clínico por sepsis, y a mediano- largo plazo la toxicidad medular. Me planteo finalmente si hubiese ofrecido un beneficio adicional realizar plasmaféresis.
Bibliografía:
-
Ordi, Ros, J; Villardel Tarrés, M. Lupus eritematoso sistémico. Farreras- Rozman. Medicina interna. Decimo sexta edición.(2008);II.;130;1101-1106.
-
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271-7.
-
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40:1725.
-
Landaw,S; Georges,J. Approach to the adult patient with thrombocytopenia. Up to date. Septiembre 2011.Versión 19.3.
-
Tristano, A. Acquired amegakaryocytic thrombocytopenic purpura: Review of a not very well-defined disorder. European Journal of Internal Medicine 16 (2005) 477 – 481.
-
Patel, M; et al. Acquired Amegakaryocytic Thrombocytopenia in a Patient With Occupational Chemical Exposure. American Journal of Therapeutics 0, 000–000 (2012).
-
Cines, D, et al. The IPT syndrome. Pathogenic and clinical diversity. Blood 2009 113: 6511-6521.
-
Ziakas,P, et al. Lupus thrombocytopenia: clinical implications and prognostic significance. Ann Rheum Dis 2005; 64: 1366-1369.
-
Levine,A; Erkan,D. Clinical assessment and management of cytopenias in lupus patients. Cuur Rheumatol Rep 2011 (13): 291-299.
-
Kuwana, et al. Autoantibody to c-Mpl (Thrombopoietin Receptor) in Systemic Lupus Erythematosus .Relationship to Thrombocytopenia With Megakaryocytic Hypoplasia. ARTHRITIS & RHEUMATISM. Vol. 46, No. 8, August 2002, pp 2148–2159.
-
Kuwana, et al. Two types of autoantibody-mediated thrombocytopenia in patients with systemic lupus erythematosus. Rheumatology 2006;45:851–854.
-
Griner, P, et al. Amegakariocytic thrombocytopenia in systemic lupus erythematosus. Arch Int Med. Vol 125. Feb 1970.
-
Agarwal, et al. Acquired Amegakaryocytic Thrombocytopenic Purpura. American Journal of Hematology 81:132–135 (2006).
-
Azuno, et al. Successful Cyclosporin A Therapy for adquired amegakariocytyc thrombocytopenic purpura. American Journal of Hematology, 69: 296-299 (2002).
-
Ueda, et al. Successful Treatment of Acquired Pure Red Cell Aplasia and Acquired Amegakaryocytic Thrombocytopenia With Anti-Thymocyte Globulin. American Journal of Hematology 66:153–156 (2001).
-
Deeren, et al. Effective use of rituximab for acquired amegakaryocytic thrombocytopenia. American Journal of Hematology (2010) Correspondence: 974-978.
-
Fukushima, et al. Successful treatment of amegakaryocytic thrombocytopenia with anti-CD20 antibody (rituximab) in a patient with systemic lupus erythematosus Lupus March 2008 vol. 17 no. 3 210-214.
-
Levine, et al. Clinical Assessment and Management of Cytopenias in Lupus Patients. Curr Rheumatol Rep (2011) 13:291–299.
-
Nguyen VA, Gotwald T, Prior C, Oberrnoser G, Sepp N. Acute pulmonary edema, capillaritis and alveolar hemorrhage: pulmonary manifestations coexistent in antiphospholipid syndrome and systemic lupus erythematosus? Lupus 2005;14(7):557-60.
-
Travis WD. Pathology of pulmonary vasculitis. Semin Respir Crit Care 2004;25(5):475-82.
-
Valente RM, Hall S, et al. Vasculitis y related disorders. In: Kelly WN, Ruddy S, editors. Textbook of Rheumatology. London: Saunders, 1997;pp:1099-10.
-
P.E Spronk, H. Boostma, G. Horst, M. G Huitema, P.C Limburg, J. W Cohen. Antineutrophil cytoplasmic antibodies in systemic lupus erythematosus. British Journal of Rheumatology 1996; 35: 625-631.
-
Manolova I, Dancheva M, halacheva K. Antineutrophil cytoplasmic antibodies in patients with systemic lupus erythematosus. Prevelence, antigen specificity, and clinical associations. Rheumatol Int 2001 Jul: 20: 197-204
-
M. Galeazzi, G. Morozzi, GD. Sebastiani, F. Bellisai, R. Marcolongo, R. Cervera, et al. Anti-neutrophil cytoplasmic antibodies in 566 European patients whit systemic lupus erythematosus: Prevalence, clinical associations and correlation whit other autoantibodies. Clinical and Experimental Rheumatology 1998; 16:541-546
-
Assicot, et al. High serum procalcitonin concentrations in patiens with sepsis and infection. The Lancet Feb 27, 1993; 341, 8884.
-
Delevaux, et al. Can procalcitonina measurement help in differentiating between bacterial infection and other kinds of inflammatory processes? Ann Rheum Dis 2003; 62:337-340.
-
Shin, et al. Serum procalcitonin measurement for detection of intercurrent infection in febrile patients with SLE. Ann Rheum Dis 2001: 60: 988-989.
|
|
|
Discusión |
|
La discusión de este seminario
corresponde al 14 de Junio de 2012, a
cargo de Dr. Francisco Consiglio |
|
| |
|
|
| |
|
|
|
|
|

